Alkyne Grasp
Reflecting work in the Fujishiro Group
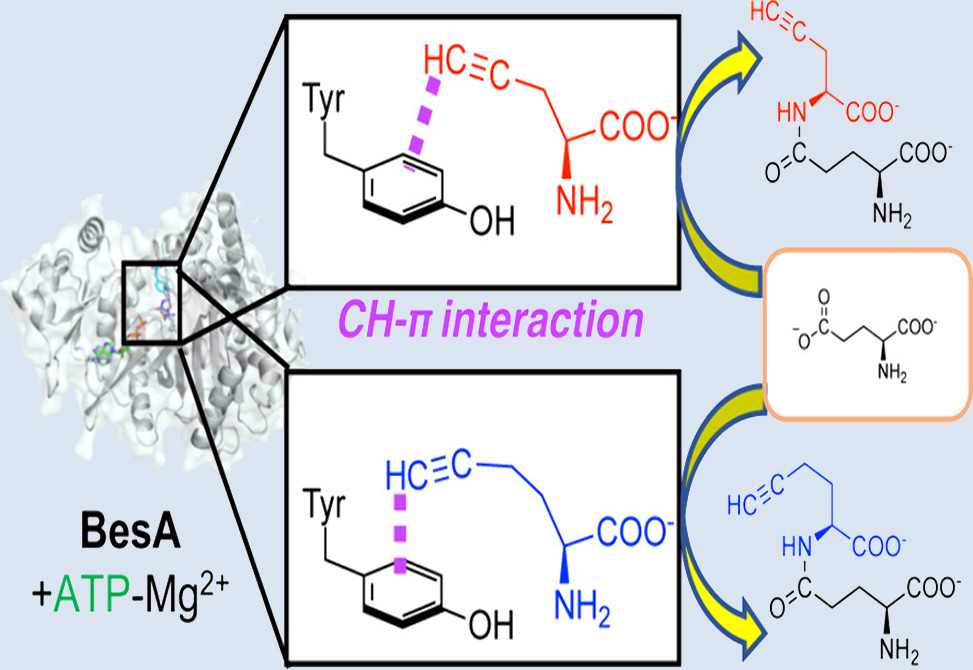
BesA is an ATP-grasp ligase that stitches L-propargylglycine to L-glutamate at the γ-position, furnishing γ-L-glutamyl-L-propargylglycine, a compact alkyne handle ideal for azide–alkyne cycloaddition. In a study published in ACS Chemical Biology, researchers in the Fujishiro Lab at the Department of Biochemistry and Molecular Biology, Graduate School of Science and Engineering, Saitama University, Japan, defines how BesA selects and positions a terminal alkyne and shows how that recognition can be repurposed to build non-natural alkyne dipeptides for click chemistry. Crystallography at 3.6 Å revealed the typical ATP-grasp fold with A, B, C, and N domains forming an ATP cavity and a narrow adjoining cleft for the amino acids. Although ATP hydrolyzed under acidic crystallization and only adenine was observed, superposition with AMP-bound ATP-grasp homologs allowed a reliable model of the nucleotide pose. Docking γ-Glu-Pra into an ADP-loaded model placed the Pra alkyne in a tight hydrophobic pocket lined by Tyr33, Ala34, and Met37, while Arg50, Arg365, and Arg404 shaped a cationic corridor for the glutamate carboxylates. The geometry positions the Pra terminal C–H toward the Tyr33 ring, consistent with stabilizing CH–π contact as the dominant alkyne recognition force, with possible minor π–π contribution.
Mutational kinetics support this picture. Wild type showed KM(L-Pra) ≈ 0.53 mM and kcat ≈ 92 min-1, in line with earlier reports. Replacing Tyr33 with Ala raised KM(L-Pra) about twenty-fold while preserving a relatively high kcat, which indicates Tyr33 governs Pra binding rather than the rate-limiting chemistry. Arg50A and Arg404A also weakened Pra binding, while Arg365A had little effect on Pra KM, which matches its distance from the alkyne in the model. Substrate handling for L-Glu differs. Wild type exhibited KM(L-Glu) ≈ 3.1 mM, the highest among tested variants, and Arg50A lowered KM(L-Glu) markedly, likely by relieving steric clash and easing entry of the γ-carboxyl toward ATP. KM(ATP) values were uniformly low, with Arg365A showing the largest increase, consistent with a role near the γ-phosphate in the ATP-bound state rather than in ADP accommodation.
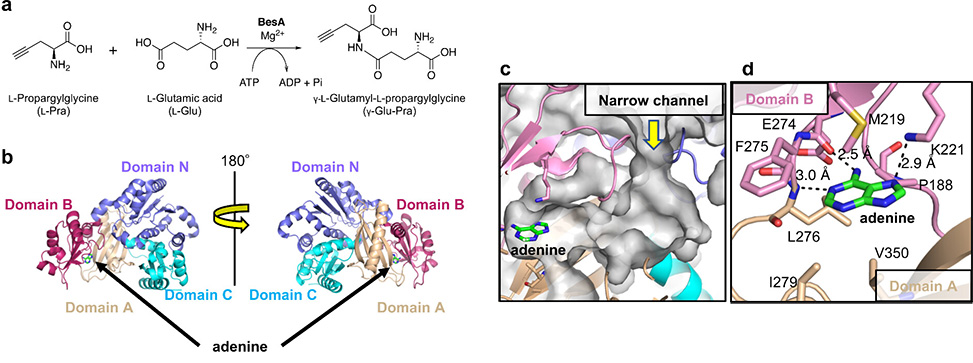
Figure 1. BesA function and structure. a| BesA-catalyzed synthesis of a terminal alkyne-containing dipeptide from natural substrates: L-propargylglycine, L-Pra, and L-glutamic acid, L-Glu. b| Two-sided views of the overall structure of BesA with adenine. Domains A, B, C, and N are highlighted in light brown, ruby red, cyan, and slate blue colors, respectively. Adenine is shown in the stick model using a black arrow. c| Surface representation of the ATP-binding site and its neighboring narrow channel and cavity for binding L-Pra and L-Glu. The surface is shown in gray. For clarity, pink has been used to denote domain B instead of ruby red. d| Surroundings of adenine. Polar interactions are shown via dashed lines.
With this structural and kinetic map in hand, the authors probed substrate scope. Only one glutamate analog, 1-methyL-L-glutamate, served as an acceptor, and only with the Arg50A variant, yielding 1-methyL-Glu-Pra at a slower rate than γ-Glu-Pra. Other analogs failed, which underscores the importance of positioning the γ-carboxyl for phosphorylation and the tight dimensional limits of the pocket. On the donor side, the enzyme accepted L-homopropargylglycine. Docking predicted that the longer alkyne still nests against Tyr33, although the terminal C–H aligns less directly. Experiments confirmed formation of γ-Glu-homoPra by wild type with a kcat only about two-fold lower than for Pra, and a higher KM that reflects looser binding. Carbon-13 labeling placed the ligation at the glutamate γ-carboxyl, the same regioselectivity observed for Pra. Variants largely lost activity with homoPra, which again highlights the fine fit around Tyr33.
These data motivate a stepwise catalytic sequence. ATP–Mg2+ binds first and is anchored by conserved polar and hydrophobic contacts in the A/B domains. Pra enters next and is captured by the Tyr33 pocket, which keeps the alkyne aligned without covalent capture. Glutamate then binds adjacent to ATP and Pra. The enzyme phosphorylates the γ-carboxyl of glutamate. The Pra amine attacks the activated carbonyl to form the γ-peptidyl linkage and release phosphate. The low KM for Pra relative to glutamate likely reflects a biological strategy in the β-ethynylserine pathway: BesA sequesters a potentially toxic Pra intermediate that can inhibit PLP enzymes, and channels it promptly into a ligated product.
The Tyr-centered CH–π clamp explains several comparative observations across alkyne-binding proteins. PLP enzymes inhibited by Pra lack an equivalent aromatic cradle, or they position Tyr for irreversible chemistry, which would be incompatible with catalysis in BesA. PutA’s preference for an alkyne over an alkene is also congruent with Tyr-mediated stabilization of a linear π system that presents a polarized terminal C–H. The model further rationalizes why L-cysteine, but not L-methionine, can masquerade as Pra in earlier screens. A sulfur–π interaction with Tyr33 is plausible for cysteine, while methionine is too bulky for the pocket.
From an applied view, BesA is already a competent biocatalyst for installing compact alkynes into dipeptides under mild conditions. Acceptance of homoPra points to routes for celL-friendly labeling since homoPra is a known methionine surrogate in translation systems. The Arg50 position emerges as a gatekeeper for the acceptor side. Substitutions that open space near the glutamate channel, combined with Tyr33 conservation for alkyne capture, should enable engineering toward bulkier or modified γ-carboxylate partners. The kinetic partitioning suggests that improving binding, not turnover chemistry, will drive gains in catalytic efficiency. Directed evolution focused on the B/N-loop cleft, with selections that reward γ-selective phosphorylation and retention of the alkyne CH–π geometry, is a logical next step.
In sum, BesA recognizes terminal alkynes through a defined Tyr33 pocket and couples that recognition to ATP-powered γ-peptide bond formation. The work provides a mechanistic foundation for tailoring BesA as a click-ready peptide ligase, expands the chemical space to include homoPra-bearing products, and clarifies how a single aromatic residue can translate a minimalist functional group into precise enzymatic selectivity.