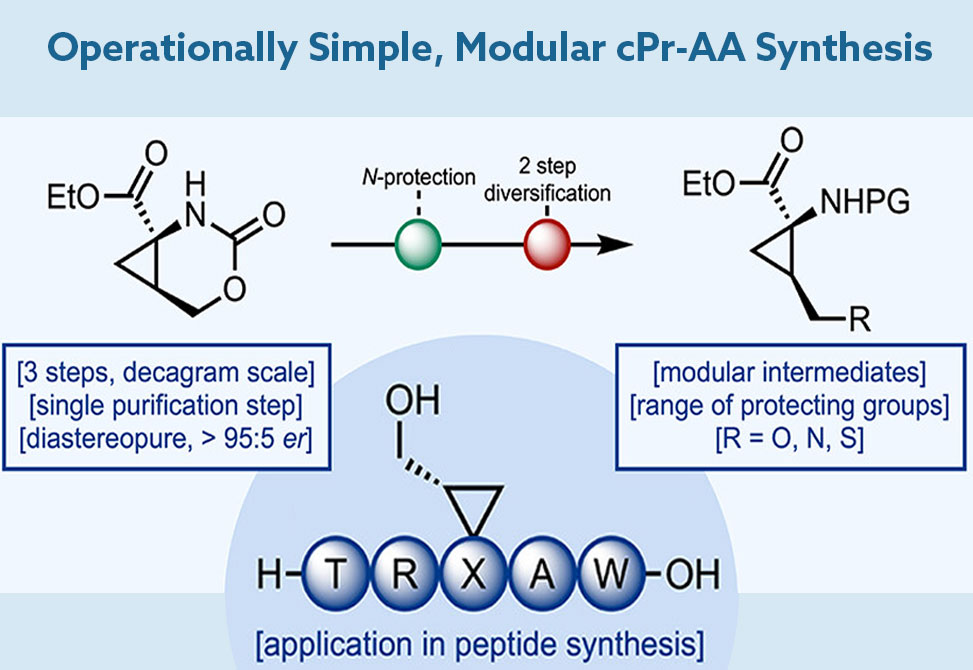
Versatile Precursors
Reflecting work in the Mitchell Lab
Cyclopropane-containing amino acids, prized for their conformational rigidity and stability, are of increasing interest in therapeutic peptide development and antimicrobial drug discovery. A recent study from Nicholas Mitchell’s group at the University of Nottingham, in collaboration with Charlie Swan under a UKRI EPSRC Doctoral Prize Fellowship, published in Organic Letters, introduces a modular, metal-free, and neurotoxin-free route to stereopure cyclopropane amino acid building blocks using common reagents and mild reaction conditions. The approach centers on a Hofmann rearrangement and cyclic carbamate intermediates, yielding versatile precursors suitable for solid-phase peptide synthesis, SPPS, and other synthetic applications.
Background
Cyclopropane motifs are widely utilized in medicinal chemistry, notably within antiviral drugs like Simeprevir and natural compounds such as coronatine. Their ring strain imparts conformational constraints that enhance receptor specificity and proteolytic resistance. However, existing routes to cyclopropane amino acids often require chiral catalysts, hazardous reagents, or laborious purification. This work seeks to streamline access to such building blocks by avoiding transition metals and neurotoxic oxidants, while maintaining high stereoselectivity.
Synthetic Strategy
The authors build upon a known malonate-epichlorohydrin pathway, enabling the generation of a key intermediate—cyclopropanecarboxamide—via a concise, chromatography-free sequence. A Hofmann rearrangement with trichloroisocyanuric acid, TCCA, facilitates intramolecular trapping of an isocyanate to generate a stable, enantioenriched cyclic carbamate. This scaffold forms the cornerstone of a versatile toolkit, allowing for various protecting group installations and downstream derivatization.
Key Advances
Scalability and Simplicity: The initial steps, involving ring-opening and amide formation, proceed cleanly on decagram scales without chromatography.
Cyclic Carbamate Stability: The resulting intermediate is chemically robust but selectively reactive, allowing nucleophilic ring-opening to yield either halogenated or hydroxylated products.
Novel Ring-Opening Bromination: A standout innovation is the SN2 bromination of protected cyclic carbamates—providing access to electrophilic intermediates without requiring alcohol activation.
Diversification via Nucleophiles: The resulting bromides undergo substitution to furnish analogues of natural residues like homocysteine and β-aminoalcohols, expanding chemical space.
SPPS Compatibility: The authors demonstrate a one-pot deprotection and Fmoc protection, yielding SPPS-ready analogues. As proof of concept, a cyclopropane-homoserine derivative was successfully incorporated into an Osteostatin analogue using standard peptide synthesis protocols.
Applications and Outlook
The synthetic strategy proves adaptable to a range of nucleophiles and oxidation pathways, producing amino acid variants relevant to drug development and peptide engineering. The cyclopropane motif’s conformational rigidity and ability to modulate biological properties make these building blocks particularly attractive for designing stable peptide therapeutics, including antimicrobial and hormone-mimicking agents.
By enabling high-fidelity, scalable synthesis of stereodefined cyclopropane amino acids from readily available starting materials, this work marks a significant step forward in expanding the noncanonical amino acid toolkit. The methodology is well-suited for modern peptide chemistry, allowing medicinal chemists to explore constrained peptide backbones without relying on rare metals or complex separations.