Ultrafast Peptide Cyclization
Reflecting work in the Sun and Xue Labs
Cyclic peptides occupy a privileged niche in chemical biology. Their constrained conformations reduce entropic penalties upon binding, impart proteolytic stability, and frequently enable access to protein–protein interaction surfaces that are inaccessible to small molecules. These attributes have made cyclic scaffolds attractive starting points for therapeutics with antimicrobial, anticancer, and anti-inflammatory activities. Yet the practical chemistry of building them has often lagged behind the imagination of their applications. Cyclization is entropically costly, and many strategies demand protecting-group gymnastics or carefully engineered monomers to avoid side reactions. The result is a field rich in approaches—disulfides, lactams, triazoles, thioethers, hydrogen-bond surrogates—but still hungry for a method that is broadly selective, gentle, and operationally simple.
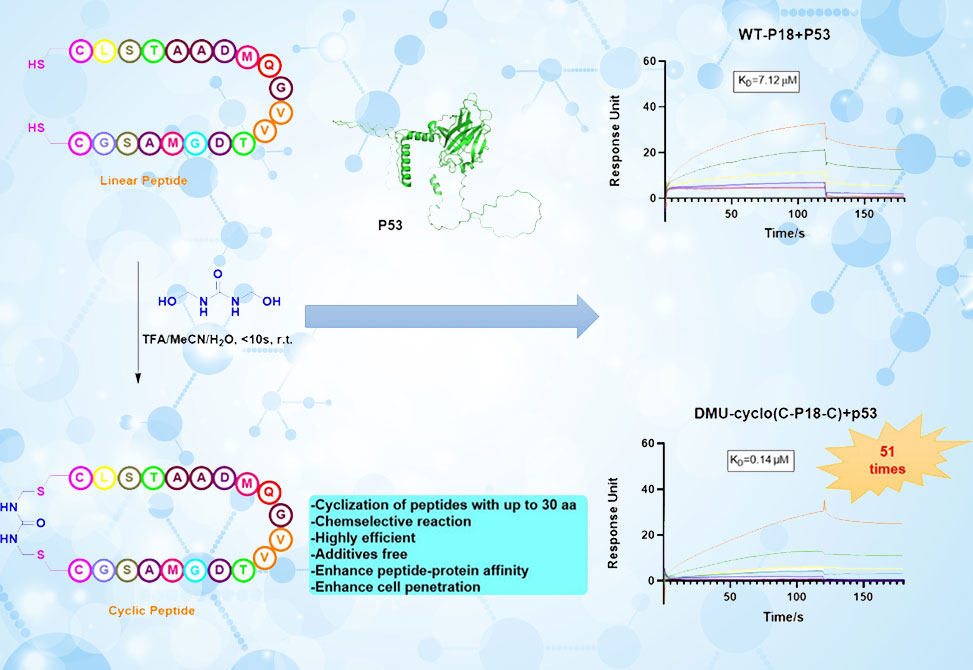
In a study published in Organic Letters, researchers from the Xue and Sun Groups, at China Pharmaceutical University and Nanjing Agricultural University, respectively, address precisely that gap by exploiting a subtle property of cysteine thiols: their nucleophilicity and low natural abundance. Cysteine has long been favored for selective conjugation, but most Cys-based cyclizations proceed under basic conditions, where competing nucleophiles—particularly amines—can intrude. Acidic conditions would be cleaner, yet few such methods exist. The authors introduce a solution: dimethylolurea, DMU, a commercially available, water-soluble reagent with two hydroxyamidomethyl groups. In the presence of trifluoroacetic acid, TFA—already ubiquitous in peptide chemistry—they show that DMU rapidly and selectively couples a pair of cysteine residues, forging a covalent bridge under conditions that are both peptide-compatible and operationally convenient.
Optimization experiments reveal the power of this approach. A model peptide cyclized with DMU in a mixed TFA/MeCN/H2O solvent undergoes >40% conversion in less than 10 seconds, and modest adjustments in solvent ratios push the yield above 70%. The reaction is insensitive to temperature, highly reproducible, and—most importantly—chemoselective: replacing the cysteines with lysines abolishes cyclization, confirming that thiols are the privileged nucleophiles. Bulky neighboring residues impose only minor penalties, underscoring the compatibility of the method with diverse sequence contexts. Even more intriguingly, the DMU-linked ring can be reopened with PdCl2, regenerating the linear precursor, providing a reversible toggle between linear and cyclic states.
The scope extends well beyond model systems. By varying the spacing between terminal cysteines, the authors prepare macrocycles of different ring sizes and observe the expected decline in efficiency with longer loops, but still detect productive cyclization even in peptides approaching thirty residues. Internal cysteine pairs also participate, yielding internally bridged macrocycles with respectable conversions. These findings suggest the method can be adapted to diverse peptide architectures, not merely terminally bracketed motifs.
Biological relevance is highlighted through two case studies. The first involves P18, an 18-mer derived from the tumor-associated protein azurin that binds p53 and prevents its ubiquitination. Cyclization of P18 via DMU preserves its α-helical structure, confirmed by circular dichroism, but dramatically enhances its affinity for p53, lowering the dissociation constant from 7.1 μM to 0.14 μM—a 50-fold improvement. The second example applies the method to pHCT74, a peptide ligand of ENO1 expressed on prostate tumor cells. Here again, cyclization improves binding, reducing the dissociation constant from 55 μM to 1.3 μM. In both cases, cyclization does not compromise folding but instead sharpens molecular recognition.
Beyond affinity, the study probes cell permeability. Fluorescein-labeled P18 derivatives demonstrate stark differences: the linear version barely enters MCF-7 cells, while the DMU-cyclized variant penetrates readily, diffusing throughout the cytosol. This underscores a broader point: cyclization not only strengthens binding but also confers the pharmacokinetic attributes necessary for intracellular targets.
Taken together, these results position DMU-mediated cysteine cyclization as an elegant addition to the peptide chemist’s toolkit. It is fast, selective, acid-tolerant, and operationally simple, leveraging reagents and conditions already familiar in peptide synthesis laboratories. The method works across ring sizes and sequence contexts, improves binding affinity in functional peptides, and enhances cell penetration—all without disturbing secondary structure. The reversibility of the linkage adds a further dimension, opening possibilities for switchable or conditionally activated peptides.
Looking ahead, the chemistry suggests fertile directions. Asymmetric DMU derivatives could install heterobifunctional crosslinks for dual functionalization. Integration into solid-phase workflows could allow “one-pot” deprotection, cleavage, and cyclization. Most importantly, the method lowers the practical barrier to exploring cyclic peptide chemical space, encouraging broader discovery of peptide modulators for targets once thought undruggable.
In short, this work reframes an old challenge with a simple insight: by moving cyclization into the acidic, TFA-dominated environment that peptides already inhabit, and by using a readily available crosslinker, one can turn cysteine’s thiol into a precise handle for macrocyclization. The result is a strategy that is as chemically neat as it is biologically consequential, offering a direct path toward peptide therapeutics with improved stability, binding, and cell access.