Copper Clicks
Reflecting work in the Metanis Lab
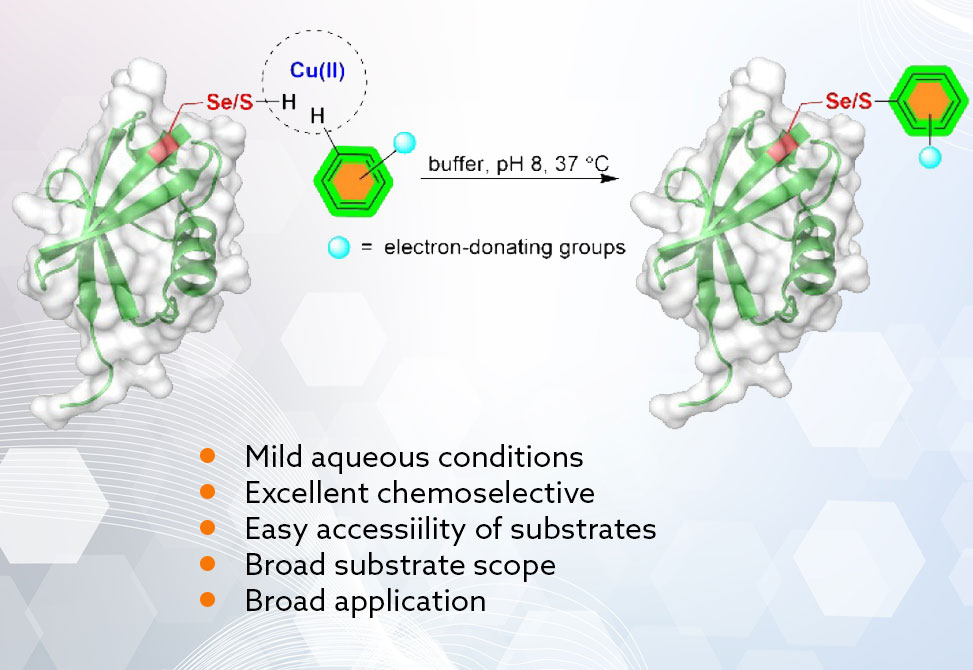
Site-specific peptide and protein bioconjugation under physiological, aqueous conditions remains a central challenge due to the dense array of competing nucleophiles and acids/bases presented in biomacromolecules. Among existing strategies, methods targeting cysteine, Cys, and selenocysteine, Sec, have become increasingly attractive because of their high nucleophilicity and low natural abundance. Yet, many strategies still require prefunctionalization of either the residue, for example, TNP activation of Sec, or the payload, for example, hydrazines, complicating workflows and risking off-target reactivity. In a collaborative work between the Metanis and Shimon labs at the Hebrew University of Jerusalem, and the Wang group at the Xiamen University in China, published in the Journal of the American Chemical Society, a copper-mediated approach is introduced that directly couples electron-rich arene C–H bonds to free S/Se–H in Cys/Sec under mild, biocompatible conditions, eliminating preinstallation steps and broadening payload scope to complex natural products and drugs. The chemistry proceeds rapidly, shows useful Sec-over-Cys selectivity that can be tuned by arene electronics, tolerates diverse sequences, and extends to peptide stapling/cross-linking and protein-level modification.
Initial screening with a Sec-containing model peptide, LGUALG-NH2; isolated as diselenide, identified Cu(OTf)2 as an efficient promoter of direct aryl–Se bond formation with resorcinol, 2a, in neutral buffer at 37 °C, delivering 93% conjugation in just 15 min. 1D/2D NMR assigned substitution at C-4 of resorcinol to give the primary monosubstituted adduct; at higher peptide loadings, di- and trisubstitution appeared, foreshadowing applications in stapling/cross-linking. Translating the conditions to a Cys analogue, LGCALG-NH2, provided a single monosubstituted product, 89% in 1 h, establishing parallel reactivity across chalcogen homologues.
Systematic variation revealed broad compatibility with resorcinol derivatives, 3b–3f, and heteroarenes including pyrroles, indoles, quinolinediol, coumarin, and pyridine. Strong electron-donating groups increased rates/yields for both Sec and Cys; in several Sec cases, disubstitution was observed, whereas Cys remained strictly monosubstituted under standard conditions—an exploitable difference for sequential chemistry. Less activated arenes, phenol, or 1,3,5-trimethoxybenzene, were inert, underscoring the need for sufficient π-donation to enter the catalytic manifold. Notably, m-phenylenediamine showed poor reactivity toward Cys despite its basicity, emphasizing that aromatic electronics, not simple nucleophilicity, govern productive coupling.
The method directly appends resveratrol, genistein, serotonin, β-carboline, harmol, vancomycin, phentolamine, and biotin, via phloroglucinol relay, to Sec-peptides with generally high conversions; serotonin and resveratrol also conjugate efficiently to Cys. This immediate access to peptide–drug conjugates, PDCs, avoids protecting-group gymnastics and enables straightforward diversification with pharmacophores spanning polyphenols, alkaloids, glycopeptides, and affinity tags.
Control peptides lacking Sec/Cys, but containing Tyr/Trp, were inert, confirming functional-group selectivity in complex sequences. MS/MS localized modification to the targeted thiol/selenol. Local environment strongly modulates outcome: Sec/Cys at termini or in open, sterically accessible contexts can yield di-adducts, two peptides per arene, whereas buried residues favored mono-adducts. This stereoelectronic sensitivity enables programmed outcomes, mono versus cross-link, by sequence design and positioning.
Using phloroglucinol, 2e, as a trifunctional hub, the team achieved high-yield stapling across Sec–Sec, 90%, Cys–Cys, 89%, with ~25% oxidized +32 Da species, and Sec–Cys, 94%, pairs in designed peptides, with tryptic mapping confirming exclusive chalcogen conjugation. Beyond intramolecular stapling, a one-pot, stepwise “Se-aryl–S” strategy cross-linked distinct peptides: first form a Cys–aryl adduct, then introduce a Sec-peptide plus fresh copper to complete intermolecular bridges. Purifying the intermediate improves final conversion, for example, 90% vs 67% for a representative pair, providing a tunable route to heterodimeric constructs. Importantly, ICP-MS of purified products showed <1 ppm residual Cu, supporting downstream biological use.
Chemically synthesized ubiquitin, (2–76)(Q2U) and Q2C variants, expressed ubiquitin(1–76)(M1S, Q2C), and Z_HER2 affibodies bearing terminal Sec or Cys were successfully modified. Highlights include 93% Sec–serotonin conjugation in 3 h, with minor deselenization over prolonged basic conditions, and ~79% Sec–vancomycin in 10 h; Cys variants gave 50–70% conversions in matched conditions, while WT controls lacking Sec/Cys were unreactive. These results demonstrate selective, functional labeling on folded proteins, extending the peptide-phase reactivity rules to macromolecular contexts of therapeutic interest, for example, HER2-binding affibodies.
Radical scavengers, TEMPO, DMPO, BHT, DPE, did not inhibit under standard aqueous conditions; however, EPR detected an aryl carbon-centered radical in the presence of Cu(II) and phloroglucinol, which was quenched by Sec/Cys peptides but not Ala. DFT supports a proton-coupled electron transfer, PCET, from the electron-rich arene to a Sec-peptide–Cu(II) complex, forming Cu(I)–Sec and an aryl radical (Ar•) that associates with Cu(I); spin delocalization onto Cu explains weak radical capture by traps. The concerted Se–Se homolysis / Se–C bond formation via the lowest barrier transition state proceeds with a ΔG‡ ≈ 19.5 kcal mol-1 and an overall exergonicity by ≈ 9.5 kcal mol-1, consistent with room-temperature kinetics. Alternate copper pathways modeled after other C–H activation proposals were less favorable. Cys follows an analogous route, with productive reactivity observed even from disulfide dimers under inert atmosphere, further aligning with the Sec mechanism.
This direct, copper-mediated arylation of free Cys/Sec furnishes a general, chemoselective, and biocompatible entry to peptide/protein conjugates without payload or residue prefunctionalization. The platform unifies: I. broad payload scope, natural products, drugs, tags; II. sequence-aware control over mono- vs di-substitution; III. efficient stapling/cross-linking architectures; and IV. protein-level labeling with sub-ppm metal residues. Mechanistic convergence on PCET-initiated, copper-bound aryl radicals provides levers—arene electronics, copper ligation, microenvironment sterics—to refine selectivity and rate.
Immediate applications include rapid assembly of PDCs, affinity/fluorophore tagging, and topology control, staples, heterodimers, in discovery workflows. Looking forward, tailoring arene electronics and ligand fields to bias Sec over Cys, or vice versa, minimizing oxidative byproducts in Cys stapling, and integrating biorthogonal handles could expand precision. The approach’s simplicity — through C–H to S/Se–H coupling in buffer — positions it as a practical workhorse for programmable bioconjugation in complex biological settings.
Publication Information
Author Information

Zhenguang was born in Shanxi, China. He received his master’s degree in organic chemistry in 2017 through a joint program between Jiangxi Normal University, where he studied under Professor Junfeng Zhao, and Nanyang Technological University in Singapore, under the supervision of Professor Chuanfa Liu. He then moved to The Hebrew University of Jerusalem to pursue his Ph.D. with Professor Norman Metanis. His doctoral research centered on advancing selenium chemistry for the chemical synthesis of complex proteins, particularly selenoproteins, and on developing Sec/Cys-specific modification strategies for functional and therapeutic applications.
In 2022, Zhenguang joined the Department of Chemistry and Chemical Biology at Harvard University as a postdoctoral fellow in the laboratory of Professor Christina Woo, where he is exploring degron chemistry to enable targeted protein degradation. Drawing on a truly international trajectory across Asia, the Middle East, and the United States, he brings together expertise in synthetic chemistry, protein design, and chemical biology to tackle some of the most challenging questions at the chemistry–biology interface.