Beyond Alpha
Reflecting work in the Yazaki Group
Unnatural amino acids have transformed both medicinal chemistry and chemical biology. By introducing side chains that nature never encoded, chemists can enhance metabolic stability, tune biological activity, and create peptide therapeutics with properties their natural counterparts lack. Among these modifications, α,β-diamino acids hold particular promise: the additional nitrogen at the β-position opens new avenues for hydrogen bonding, metal coordination, and further derivatization. Yet installing that nitrogen directly remains extraordinarily difficult. While transition-metal-catalyzed C–H activation has revolutionized how chemists think about molecular editing, β-C–H amination of amino acid derivatives has resisted general solutions. Previous approaches required intramolecular reactions that delivered β-lactam products, useful but limited in scope and requiring harsh conditions for further manipulation.
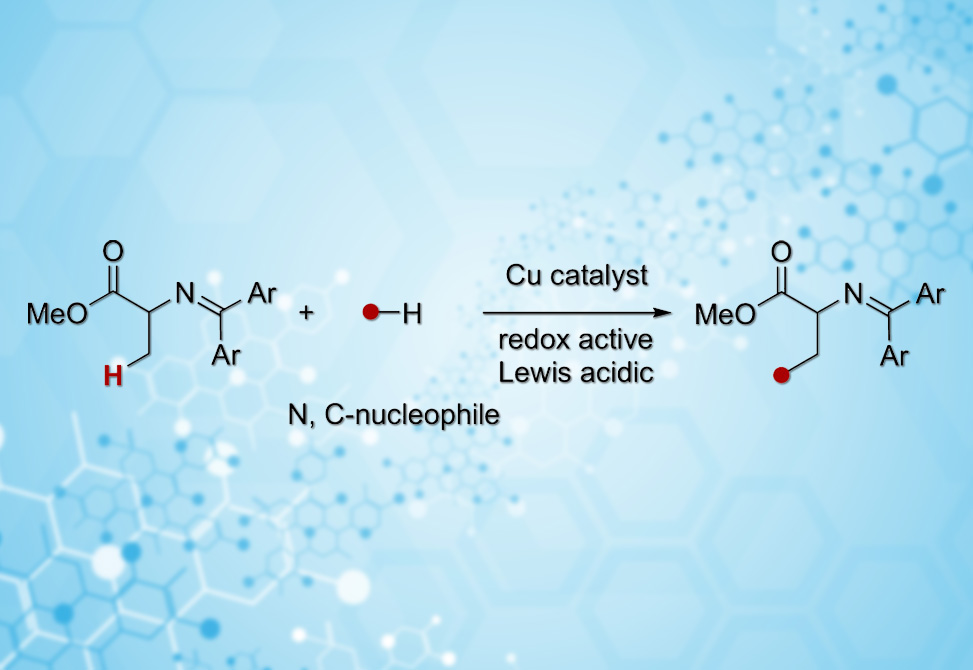
Researchers under the guidance of Takashi Ohshima and Ryo Yazaki at Kyushu University, publishing in Organic Letters, recognized that amino acid Schiff bases might hold the key. These readily available building blocks have served synthetic chemists for decades, but exclusively for α-functionalization through enolate chemistry pioneered by O'Donnell. The Japanese team reasoned that under the right conditions, a transiently formed azaallyl radical could undergo hydrogen atom transfer at the β-position, generating an α,β-unsaturated intermediate primed for conjugate addition by an amine nucleophile.
Realizing this vision required identifying a reagent that could serve dual roles: abstracting the β-hydrogen and reoxidizing the copper catalyst to sustain turnover. After screening various oxidants, the team found that TEMPO, 2,2,6,6-tetramethylpiperidine-1-oxyl, delivered both functions elegantly. Copper acetate with the ligand bathophenanthroline completed the catalytic system, though optimization revealed an unexpected subtlety. Equimolar ligand-to-copper ratios gave poor reproducibility. Detailed mechanistic studies explained why: the copper catalyst must operate in two distinct modes during the catalytic cycle. Ligand-bound copper acts as a cooperative Lewis acid and Brønsted base, deprotonating the α-position to generate an enolate that equilibrates with the key azaallyl radical. After TEMPO-mediated hydrogen atom transfer produces the unsaturated intermediate, a different copper species takes over. Ligand-free copper acetate, more Lewis acidic than its ligand-bound counterpart, promotes the conjugate addition of the amine nucleophile. Using just 7 mol% bathophenanthroline relative to 10 mol% copper acetate struck the optimal balance, delivering products in up to 90% yield.
The substrate scope proved impressively broad. Cyclic amines from four- to seven-membered rings reacted efficiently, including morpholine, piperazine, and piperidine derivatives bearing acid-sensitive acetals. Spirocyclic amines, increasingly valued scaffolds in medicinal chemistry, coupled without difficulty. Even challenging acyclic amines participated, albeit in moderate yields. Most compellingly, the method tolerated complex pharmaceutical compounds as coupling partners. Trimetazidine, troxipide, amoxapine, desloratadine, and a donepezil derivative all delivered β-aminated products in 45–85% yield, demonstrating remarkable functional group compatibility. The transformation also extended beyond amination: dibenzyl malonate underwent β-alkylation under similar conditions, pointing toward broader applications.
By repurposing amino acid Schiff bases for β-functionalization, this work breaks a longstanding limitation in amino acid chemistry. The dual catalytic mode, switching between ligand-bound and ligand-free copper species within a single cycle, offers a mechanistic blueprint that may inspire related transformations. For peptide chemists seeking direct routes to α,β-diamino acid building blocks, this copper-catalyzed approach delivers both efficiency and versatility.