Nitrogen Walks
Reflecting work in the Labs of Xiao & Huang
Noncanonical amino acids have become indispensable tools in chemical biology and drug discovery. By introducing side chains that nature never encoded, researchers can endow peptides and proteins with new functions, improved pharmacokinetics, and enhanced material properties. While chemical synthesis delivers these building blocks efficiently, biocatalytic routes offer compelling advantages in selectivity and sustainability. Established enzymatic strategies include transamination, electrophilic addition using tryptophan synthase, and late-stage functionalization with hydroxylases or halogenases. Yet most of these methods rely on native enzyme mechanisms. Importing abiotic reaction pathways into enzyme active sites could dramatically expand the biocatalytic toolbox, combining the genetic tunability of proteins with the mechanistic diversity of synthetic chemistry.
A team led by Xiongyi Huang and Han Xiao, publishing in the Journal of the American Chemical Society, recognized that nonheme iron enzymes possess uniquely flexible coordination environments suited to such unnatural transformations. They drew inspiration from recent work showing that chiral ruthenium and iron catalysts bearing two cis-labile coordination sites can promote enantioselective amino acid synthesis through 1,3-nitrogen migration. In this mechanism, an iron-nitrenoid intermediate abstracts a hydrogen atom from a nearby carbon, and the nitrogen then migrates to that position. The team hypothesized that the open active site of a nonheme iron enzyme could enable this same pathway while providing the chiral environment necessary for stereochemical control.
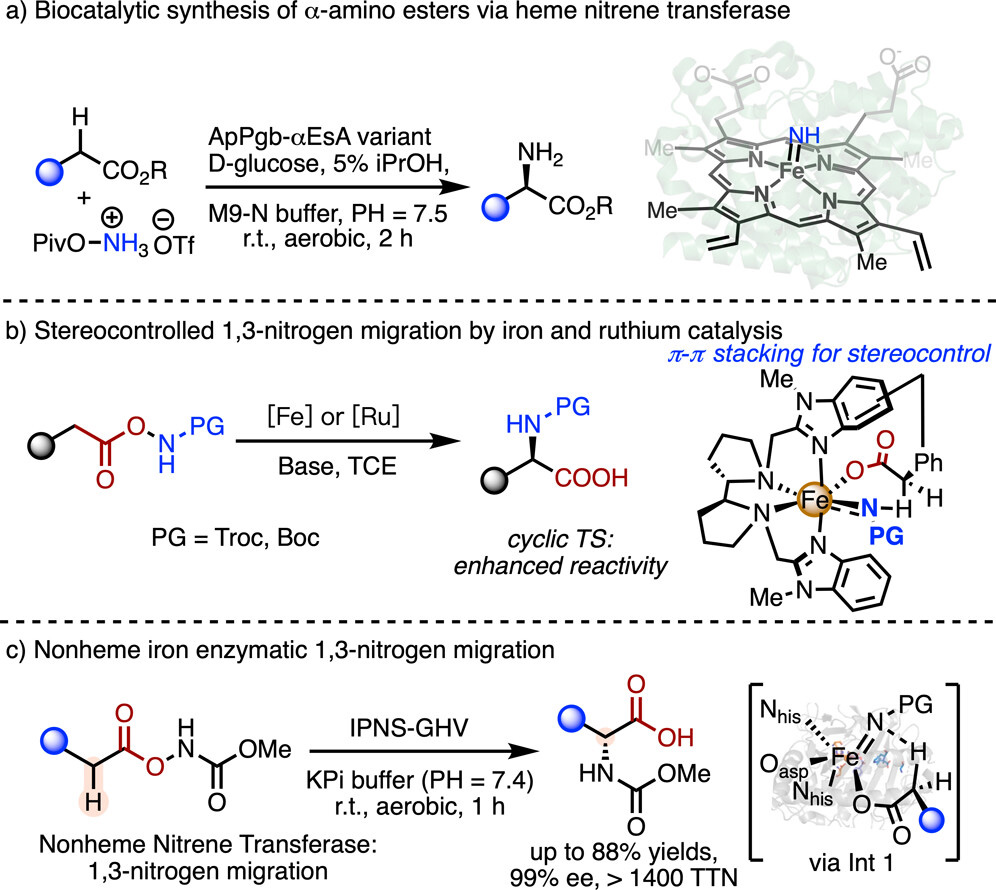
Figure 1. Chemical and biocatalytic synthesis of ncAA derivatives via C–H amination.
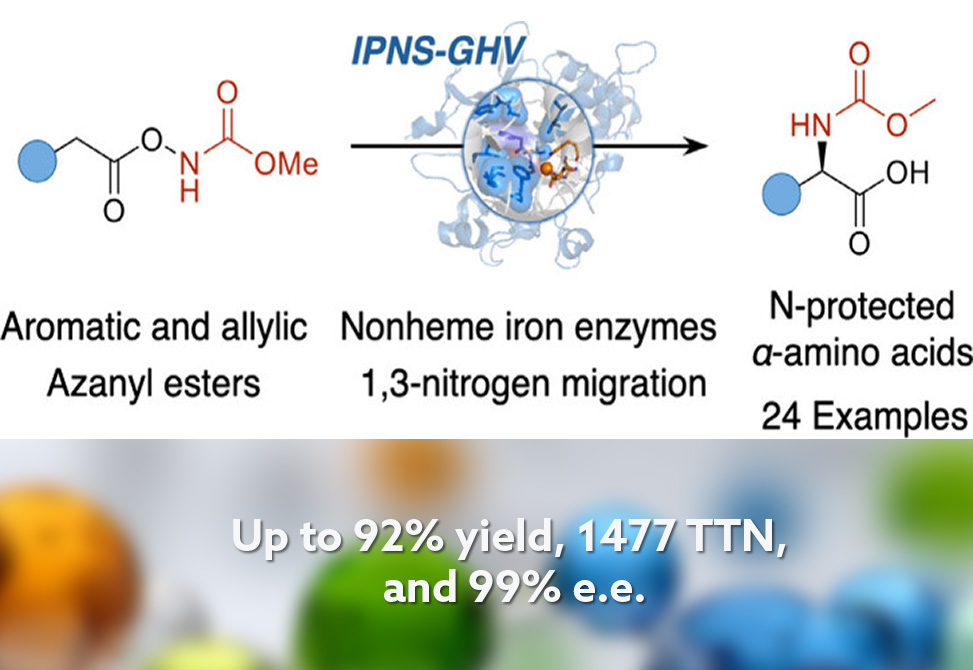
Screening a library of nonheme enzymes against an azanyl ester substrate identified isopenicillin N synthase, IPNS, from Aspergillus nidulans as a promising starting point, delivering product in 4% yield. This represents the first demonstration of IPNS catalyzing a non-natural transformation. Conducting reactions under anaerobic conditions improved yield to 22% with 83% enantiomeric excess. Control experiments confirmed the essential role of iron: replacing the metal center or mutating the iron-coordinating histidine residues abolished activity. The methyl carbamate protecting group on the nitrogen precursor proved optimal, distinguishing this system from a contemporaneous report using a different nonheme scaffold that favored tert-butyloxycarbonyl protection.
Docking simulations guided directed evolution. Three rounds of iterative site-saturation mutagenesis at active-site residues yielded IPNS-GHV, a triple mutant that produced the D-phenylglycine product in 72% yield and 98% enantiomeric excess with a total turnover number exceeding 3000. Molecular dynamics simulations confirmed that these mutations preserved overall protein stability while reshaping the reactive pocket. The optimized system tolerated electron-rich and electron-deficient phenylacetic acids, heteroaromatic substrates, and para- and meta-substituted derivatives, all with excellent enantioselectivity. Ortho-substitution reduced yields modestly due to steric constraints. To address allylic substrates, two additional evolution rounds produced IPNS-GHCGV, a pentuple mutant achieving 63% yield and 91% enantiomeric excess for allylic C–H amination. Three products were prepared on millimole scale, demonstrating practical utility.
Mechanistic studies supported a radical pathway. A kinetic isotope effect of 2.1 indicated that C–H bond cleavage contributes to the rate-determining step. The radical scavenger TEMPO completely suppressed the reaction, and TEMPO-trapped intermediates were detected. Substrates lacking the carboxylate tether gave no product, confirming that chelation to iron and preassociation between substrate and nitrene precursor are essential for reactivity. Compared to small-molecule iron catalysts that achieve similar transformations, this biocatalytic platform avoids halogenated solvents and delivers roughly 200-fold higher turnover numbers. By repurposing IPNS for chemistry far removed from its native penicillin biosynthesis, this work expands the frontier of new-to-nature enzymology for amino acid synthesis.