Short linear motifs, SLiMs, encode compact regulatory instructions across the proteome, and their recognition by peptide-binding domains such as SH2 modules underlies the wiring of major signaling circuits. Because SLiM-PRD interfaces are acutely sensitive to local sequence context, single amino-acid substitutions can reconfigure interaction networks, enabling evolutionary innovation but also fostering pathogenic rewiring. Yet despite decades of progress, accurate quantitative prediction of SH2–SLiM binding affinities has remained elusive. Classical position-specific scoring matrices and modern discriminative classifiers readily identify candidate sites, but they fail to reproduce biophysically meaningful free-energy landscapes. In this context, a collaborative study from the Bussemaker and Shah Groups at Columbia University, published in Protein Science, establishes an integrated experimental–computational framework, built on massively diverse peptide-display libraries, multi-round affinity selection, next-generation sequencing, and the ProBound free-energy regression method, that yields quantitative, domain-specific sequence-to-affinity models capable of predicting binding free energies across the full theoretical ligand space.
The strategy is validated across diverse library designs, ranging from semirational pTyrVar collections to fully degenerate random libraries. Importantly, the authors demonstrate that even unbiased X11 libraries—whose naïve state is dominated by weak binders and low-frequency sequences—can be transformed into informative training data through carefully optimized multi-round selection, recovering free-energy parameters that agree closely with models derived from more constrained library formats. This universality is significant. It allows a single random library to serve as a target-agnostic substrate for profiling entirely new peptide recognition domains without relying on prior knowledge of binding motifs. Furthermore, by removing constraints on the central tyrosine and still recovering coherent binding models, the approach proves capable of discovering binding determinants without imposing motif-specific assumptions.
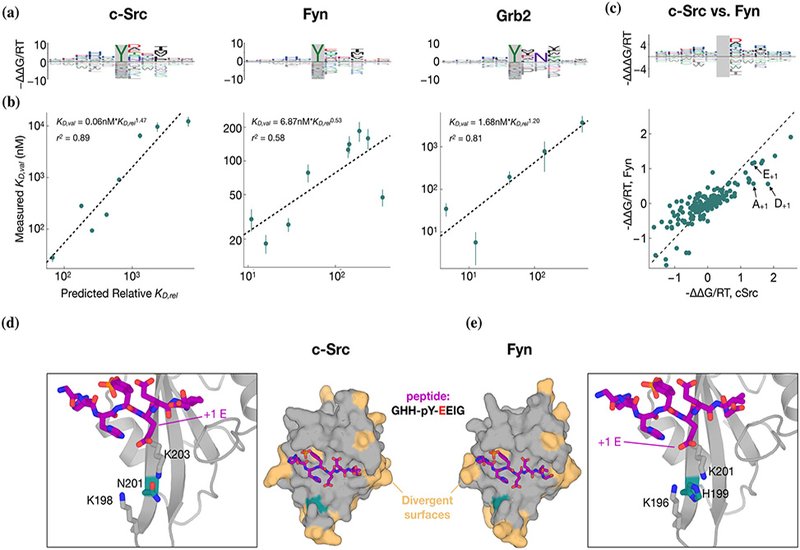
Quantitative accuracy is established via rigorous low-throughput biophysical validation. Competitive fluorescence polarization measurements across panels of natural and engineered phosphopeptides reveal strong agreement between predicted and measured affinities, spanning nearly two orders of magnitude in KD. For Grb2, the hallmark requirement for an asparagine at +2 emerges naturally and is resolved quantitatively: peptides bearing N+2 are predicted, and observed, to bind with affinities that differ by several orders of magnitude relative to N+2 mutants. Likewise, nuanced paralog-specific preferences distinguishing c-Src and Fyn are illuminated, and structural modeling suggests that subtle divergence at a single conserved pocket residue, N201 in c-Src vs. H199 in Fyn, reshapes the electrostatics of the pY+1 binding cleft, rationalizing experimentally observed differences in affinity for glutamate, aspartate, or alanine at that position.
Armed with validated affinity models, the authors extend their analysis to the human tyrosine phosphoproteome, assigning relative affinities to thousands of known phosphosites while filtering for physiologically plausible co-expression. The resulting predictions identify candidate SH2 interactors—including several not previously reported in curated interaction databases, and the model’s ability to discriminate known binding vs. non-binding phosphopeptides in external datasets further supports its generality. The framework is also applied to single-amino-acid variants from clinically relevant mutation databases; in multiple cases, predicted gains or losses of affinity align with known pathogenic signaling mechanisms, offering mechanistic insight into allelic rewiring events involving Vav2, ZAP-70, HS-1, and ARPC1B.
Overall, the study demonstrates that quantitative sequence-to-affinity modeling of SH2 domains is achievable directly from high-throughput peptide display data, without requiring explicit affinity measurements. By accurately reconstructing free-energy landscapes from raw sequencing counts and capturing paralog-specific specificity determinants, this approach offers a powerful route to deciphering SLiM-mediated interaction networks at proteome scale. More broadly, it establishes a generalizable blueprint for mapping binding energetics across diverse peptide-recognition domains, moving beyond binary classifications toward predictive, energetically grounded models of cellular signaling fidelity and its perturbation by natural or disease-associated variation.