News Archive


From Moth to Medicine
Omnix Medical has advanced OMN6, a cyclized, protease-resistant antimicrobial peptide engineered from the classic insect peptide Cecropin A, into Phase II trials against carbapenem-resistant Acinetobacter …

Helma Wennemers Receives the Emil Fischer Medal
The German Chemical Society will award Prof. Helma Wennemers the Emil Fischer Medal, its historic prize for organic chemistry, at the ORCHEM conference in Freiburg …

In Memory of Dr. Richard Houghten
On July the 5th, the peptide and drug-discovery communities lost one of their greatest pioneers. Dr. Richard Houghten was a visionary scientist whose innovations transformed …

A Foundation for Design
The Novo Nordisk Foundation Center for Protein Design has just moved into its new labs and offices at the University of Copenhagen, with an official …

Heinis Earns ERC Advanced Grant
Professor Christian Heinis has won a 2025 ERC Advanced Grant from the European Research Council. Heinis, who heads the Laboratory of Therapeutic Proteins and Peptides …
Call for Papers
Radiopharmaceuticals have emerged as a rapidly evolving field at the interface of medicinal chemistry, molecular imaging, and targeted therapy. Progress in radiopharmaceutical development and translational …

Kiessling and Raines Recognized
The American Peptide Society congratulates Laura L. Kiessling and Ronald T. Raines of MIT on receiving the 2026 van 't Hoff Lectureship Award from the …
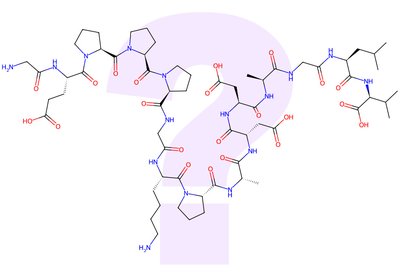
The BPC-157 Question
A fifteen-amino-acid peptide named BPC-157 has spent fifty years on the margins of medicine and is now headed for an FDA advisory review. In a …

Call for Papers
This Special Issue of OPR&D; will highlight advances in design, synthesis, purification, and manufacturing strategies that translate molecular complexity into process reliability. Submit your manuscript …


Helma Wennemers Receives the Wilhelm Ostwald Medal
Prof. Helma Wennemers is one of two recipients this year of the Wilhelm Ostwald Medal, the Saxon Academy of Sciences' most important prize, awarded in …