Tuberculosis, TB, remains one of the most pressing global health threats, with more than ten million cases reported in 2022. Once thought to be in gradual decline, TB incidence has reversed course, fueled by the rise of multidrug-resistant, MDR, and extensively drug-resistant, XDR, strains. The causative pathogen, Mycobacterium tuberculosis, Mtb, owes much of its persistence to its formidable cell envelope — a multilayered barrier comprising an inner membrane, peptidoglycan, an arabinogalactan layer, and, most critically, an unusually thick, hydrophobic mycomembrane. Rich in mycolic acids and glycolipids, this outer layer forms a permeability barrier that has shaped the chemical space of effective TB drugs. Nearly all frontline antibiotics are small, hydrophobic molecules that can squeeze through this envelope, but decades of overuse have left them vulnerable to resistance. Only two new drugs, bedaquiline and pretomanid, have been introduced in recent years, both still small and hydrophobic.
Yet there are striking exceptions. Rifampicin and streptomycin, for instance, show that larger, more polar molecules — including peptides — can in fact succeed against mycobacteria. The pharmaceutical field more broadly has already embraced peptides, particularly those exceeding Lipinski’s Rule of Five, as promising scaffolds in oncology, metabolic disease, and antibacterial discovery. Macrocyclic peptides such as Zosurabalpin, which targets carbapenem-resistant Acinetobacter baumannii, underscore the potential of these scaffolds in otherwise intractable infections. Natural products like evybactin and cyclomarin A have further fueled optimism, inspiring BacPROTAC-based antimycobacterial strategies.
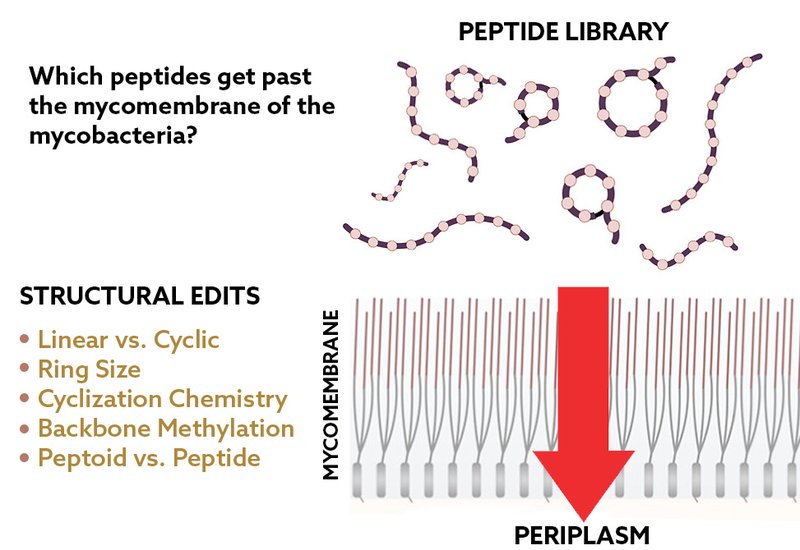
A key bottleneck in mycobacterial peptide development, however, has been the inability to measure whether candidate molecules truly accumulate past the mycomembrane. Standard LC–MS/MS can quantify whole-cell associations but cannot readily pinpoint subcellular location. To address this, researchers in the Marcos Pires Group at the University of Virgina, published in ACS Chemical Biology, under first authors Rachita Dash and Zichen Liu, developed PAC-MAN — Peptidoglycan Accessibility Click-Mediated Assessment — a live-cell assay that uses engineered peptidoglycan landmarks, via DBCO modification, and bioorthogonal click chemistry to "register" the arrival of azide-tagged molecules at the PG layer. In essence, PAC-MAN reports directly on whether a molecule has traversed the mycomembrane, offering both throughput and precision at the periplasmic scale.
Using Mycobacterium smegmatis as a model system, the researchers optimized PAC-MAN for peptides, establishing dose-dependent accumulation curves that paralleled mammalian penetration assays such as Chloroalkane Penetration Assay, CAPA. They validated the chemical installation of dibenzocyclooctyne, DBCO. in the peptidoglycan, confirmed that fluorophore choice and labeling concentrations did not confound results, and showed that apparent accumulation correlated robustly with exposure time and concentration.
With this foundation, they turned to two structural strategies common in natural products but not empirically tested in mycobacteria: backbone N-alkylation and macrocyclization. Backbone N-methylation of simple model peptides revealed that just one or two methyl groups could sharply enhance accumulation, particularly toward the C-terminal backbone. Higher levels of methylation did not always confer added benefit, and in some cases reduced accumulation, reflecting the nuanced balance between conformational preferences and permeability. Extending this logic, the team built peptoid analogs, which replace backbone amides with N-substituted glycines. These peptoids often accumulated even more effectively than methylated peptides, highlighting the broad potential of backbone alkylation to reduce desolvation penalties and promote passage through the mycomembrane.
Macrocyclization offered complementary insights. Cyclic peptides consistently outperformed their linear analogs, especially at smaller ring sizes where intramolecular hydrogen bonding reduces solvent exposure. Lariat-shaped macrocycles showed organism-specific trends, with some scaffolds accumulating better in M. tuberculosis than in M. smegmatis. Cyclization chemistry also mattered: thioether-linked cycles improved accumulation, while disulfide linkages sometimes introduced vulnerabilities to thiol exchange at the envelope. NMR hydrogen–deuterium exchange experiments confirmed that cyclic peptides exhibited greater protection of backbone amide hydrogens, consistent with reduced solvent accessibility. Moreover, cyclic scaffolds proved far more stable in serum than their linear counterparts, underscoring the pharmacological benefits of cyclization.
The team then applied these principles to real antibiotic scaffolds. Tridecaptin A1, traditionally inactive against mycobacteria, was redesigned through N-methylation and macrocyclization. These modifications improved both accumulation and antimicrobial activity, reducing MIC values against M. smegmatis by up to eight-fold. Interestingly, while the parent tridecaptin disrupted the cell envelope nonspecifically, N-methylated analogs avoided this liability while gaining potency. Griselimycin, a natural product already bearing both macrocyclization and N-methylation, provided a complementary test: removing either feature significantly reduced activity, confirming their essential roles in preserving efficacy.
Together, these results provide the first systematic rules for tuning peptide permeability in mycobacteria. N-alkylation and macrocyclization — long recognized in mammalian systems — also enhance peptide accumulation across the mycomembrane, though with important context-dependent subtleties. PAC-MAN enables these relationships to be mapped at scale, establishing empirical guidelines for redesigning natural products and synthetic peptides alike.
This work reframes the conventional belief that mycobacteria are impenetrable to large, hydrophilic molecules. Instead, it demonstrates that rational structural edits can transform poorly permeable peptides into viable antimycobacterial candidates. By aligning natural product-inspired chemistry with empirical accumulation data, Dash and colleagues have laid the groundwork for a new peptide-based antibiotic pipeline against TB and its drug-resistant forms.