Coagulation Trigger
Reflecting work in the Ruvo and Sandomenico Labs
Hemostasis begins with a handshake. When vascular damage exposes tissue factor, TF, to the bloodstream, factor VII, FVII, latches on, instantly engaging the extrinsic pathway and accelerating thrombin generation. Because TF is sequestered outside intact vessels, the TF:FVIIa complex behaves like a conditional switch: silent in health, fast and local in injury. That logic makes the interface a compelling drug target—intercept the switch and you can temper coagulation before the system amplifies itself, potentially with less bleeding risk than blocking catalytic sites downstream.
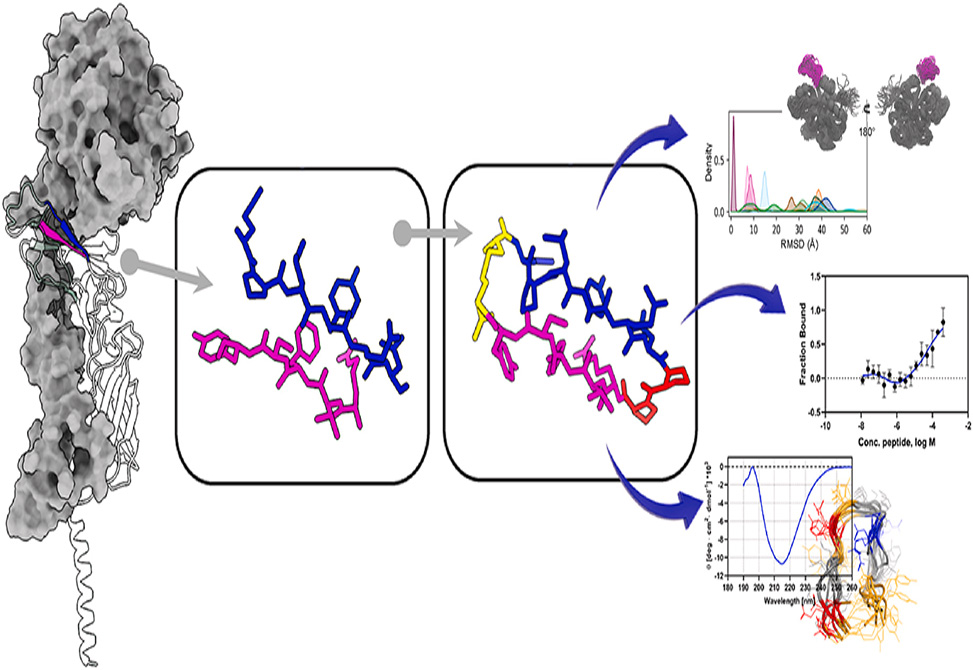
In work published in the European Journal of Medicinal Chemistry, researchers from the Institute of Biostructure and Bioimaging, Naples, Department of Molecular Medicine, Sapienza University of Rome, University Federico II, Naples, and BIOVIIIx, Naples, Italy, pursue an elegant minimalism: distill a sliver of TF’s β-sheet surface that grips FVIIa, and present it back to the protease as a compact β-hairpin. The structural template comes from the crystal complex, for example, PDB 1DAN/2A2Q, where two TF strands—106–110, RVFSY, and 123–128, EPLYEN, abut a narrow hot spot on FVIIa centered on Met366 within the LMTQD, 365–369 segment. To translate that interface into a peptide, they tether the two TF strands with a di-proline turn and staple the termini with a disulfide, yielding a monocyclic scaffold with the general architecture Cys–EPLYEN–Pro–Pro–RVFSY–Cys. Crucially, the chirality of each proline is a design dial. By permuting L/D configurations—PP, Pp, pP, pp—they can bias the turn geometry and, with luck, recreate the strand alignment TF uses in the native complex.
The chemistry performs cleanly. Linear precursors cyclize in ~16 h to >95% purity with masses matching theory; yields settle around 20–29%. Recombinant human FVII, HEK293 expression, behaves as expected by SDS–PAGE, CD, and a chromogenic activity assay, providing a reliable binding and function partner.
Conformation proves to be destiny. CD spectroscopy at neutral pH draws a line between candidates: pP-TF, D-Pro–L-Pro, shows a strong β-sheet signature, minimum ~215 nm, pp-TF, D–D, shows a weaker but present sheet signal, while PP-TF, L–L, and Pp-TF, L–D, look random. NMR sharpens the picture. All four peptides display cis/trans behavior at the central proline–proline bond, but the populations differ: pP-TF is ~70% trans and adopts a canonical β-hairpin in the majority of structures; pp-TF achieves a β-hairpin only in its ~25–30% cis subpopulation; PP-TF and Pp-TF distribute among multiple isomers with no stable hairpin. The spectroscopic hierarchy foreshadows biology.
When binding is quantified by microscale thermophoresis, only the hairpin-competent peptides engage FVII: pP-TF and pp-TF bind in the low-to-mid micromolar range, K_D ≈ 70 ± 25 μM and 67 ± 20 μM, respectively. The random-coil variants do not register measurable affinity. Function tracks structure as well. In a TF-dependent chromogenic assay, pP-TF suppresses FVII activity by >40% at 50–100 μM; pp-TF achieves ~11% and ~27% inhibition at the same doses; Pp-TF barely moves the needle, ~13% at 100 μM. For short, rigid scaffolds hand-picked from an extended protein interface, those are meaningful effects at an exceptionally upstream checkpoint in the cascade.
Computation fills in the contact physics. All-atom MD of each cyclopeptide docked onto FVIIa begins from the TF-derived pose and watches what holds. pP-TF stays where it should: its RMSD centers around ~2.2 Å, it preserves salt bridges between Glu2 and Arg364/Arg439 and between Arg10 and Asp369, and it intermittently forms π–π contacts between Phe12/Tyr14 and Phe335 on FVIIa. Contact maps echo the experimental design: pP-TF most faithfully recapitulates the TF segments’ interactions with the FVIIa 365–369 hotspot and neighboring R331/F335/M336–D339/R439 region. pp-TF, less pre-organized, samples multiple surface patches and often migrates toward a flexible loop, 189–210. PP-TF and Pp-TF disperse further, with contacts transient and poorly aligned to the native TF footprint. A residue-level RMSF analysis mirrors those trends: pP-TF is the least mobile overall, while Tyr5, Arg10, Phe12 are universally the liveliest side chains, consistent with their roles at the binding interface.
Two design choices deserve special credit. First, stereochemical tuning at the turn: flipping one proline to D enforces the geometry needed for the strands to face each other and register hydrogen bonds—hence the leap in β-hairpin population, and activity, for pP-TF versus PP-TF. Second, placing the rigid element, the Pro–Pro linkage, at the closer ends of the native strands, N128↔R106, and the more flexible disulfide across the farther ends, E123↔Y110, reproduces the spatial constraints seen in TF, rather than fighting them. Together those moves convert a broad, multi-contact protein surface into a 13–14 residue loop that actually behaves like the parent interface.
How do these hairpin mimics stack up pharmacologically? They will not out-compete active-site protease inhibitors in potency. But potency is not the point. These synthetic, conformationally defined cyclopeptides strike at the TF:FVIIa handshake itself, a regulatory move that preserves downstream protease function elsewhere and may therefore lower bleeding liability relative to catalytic blockade. Compared with antibodies or recombinant inhibitors, for example, TFPI, active-site inhibited FVIIa, they are smaller, cheaper to make, and structurally modular. Compared with earlier loop-derived peptides from TF or FVII, often millimolar binders, they are both tighter and better organized. And because their activity correlates tightly with β-hairpin content, the path to optimization is clear: stabilize the hairpin, strengthen the hot-spot contacts, and maintain the disulfide- and turn-encoded rigidity that underwrites enzymatic stability.
That roadmap is already sketched. Turn surrogates could replace the di-Pro to fix the geometry and resist isomerization. Targeted side-chain edits on the RVFSY/EPLYEN faces, for example, electrostatic reinforcement of the Glu2↔Arg364/Arg439 and Arg10↔Asp369 axes or aromatic tuning around Phe335, should boost on-target residence. Depeptidizing the flanks while retaining the disulfide and hairpin hydrogen-bonding could improve exposure without sacrificing fold. And the design space is not limited to FVIIa: hairpin grafts can be transplanted onto heterogeneous scaffolds to tune PK while preserving the interaction grammar learned here.
The biological logic remains compelling. Because TF lives outside intact vessels, interfering with the TF:FVIIa interface promises context-dependent anticoagulation—quieter in health, more active where TF is pathologically exposed, for example, DIC, sepsis, cancer-associated coagulopathy. In that sense these β-hairpin mimics are not merely binders; they are structurally informed levers at the top of the coagulation cascade. With the pP-TF scaffold as a starting point—conformationally compact, β-hairpin-rich, and already functional—the next generation of TF-interface inhibitors looks less like a moonshot and more like a matter of precision stereochemistry and contact engineering.