The Ras superfamily of small GTPases drives cell growth, migration, and cytoskeletal remodeling, making these proteins attractive cancer targets. Covalent inhibitors of K-Ras G12C have reached the clinic, but resistance mechanisms emerge rapidly, and many oncogenic GTPase variants lack the reactive residues needed for covalent targeting. Small GTPases as a class present a fundamental challenge: their protein surfaces lack the deep pockets that small molecules typically exploit. Many Cdc42 effector proteins bind by extending a beta-strand alongside the GTPase's own beta-sheet, creating a flat, expansive interface that has long resisted conventional drug design. Developing inhibitors that can compete with these natural protein partners at such featureless surfaces remains a frontier in therapeutic targeting of the Ras superfamily.
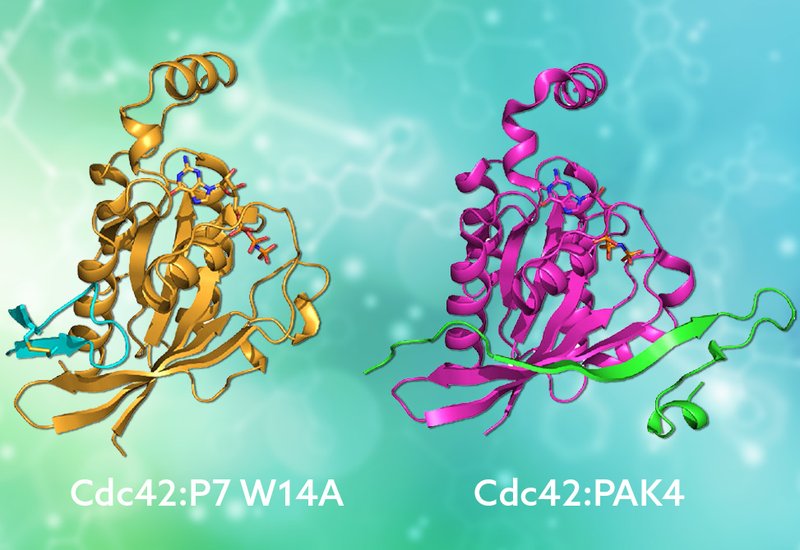
Researchers supervised by Drs. Helen Mott and Darerca Owen at the University of Cambridge, published in Biochemistry, in collaboration with Dr. Jefferson Revell at AstraZeneca, engineered a new generation of disulfide-cyclized peptide inhibitors that target Cdc42 by mimicking the binding mode of its native effector proteins. Building on their earlier second-generation peptide P7, which bound Cdc42 with 24 nanomolar affinity and inhibited proliferation and migration in Ras-driven cancer cells, the team employed systematic alanine scanning to map the energetic contribution of each residue. A single substitution, replacing tryptophan-14 with alanine, improved affinity nearly four-fold. Using a combination of NMR experiments, the team then solved the solution structure of this optimized peptide, designated P7 W14A, in complex with Cdc42.
The structure revealed a striking result: the peptide adopts an ordered beta-hairpin conformation and docks against the beta-2 strand of the GTPase Rossmann fold, forming a short antiparallel beta-sheet that extends the protein's own secondary structure. This binding mode directly mimics how natural CRIB domain effectors like PAK, WASP, and ACK engage Cdc42, with a conserved isoleucine in the peptide occupying the same hydrophobic pocket used by the native effector isoleucine. Remarkably, this mimicry emerged from a peptide originally selected from a fully random library, not from rational design based on effector sequences. NMR structures of the free peptide showed that the beta-hairpin forms only upon binding, transitioning from a loose, unstructured loop to the ordered conformation through conformational selection. Guided by the bound structure, the team then designed targeted substitutions at the binding interface and within the peptide scaffold. Replacing histidine-7 with tyrosine improved affinity 7.5-fold, while incorporating a noncanonical 5-bromotryptophan at position 11, predicted to form a halogen bond with a neighboring lysine on Cdc42, delivered a 9-fold enhancement. Structural substitutions using 2-aminoisobutyric acid were well tolerated and could offer improved proteolytic stability, an important feature for therapeutic development.
This work demonstrates that the flat, seemingly undruggable effector-binding surfaces of small GTPases can be selectively targeted by constrained peptides that recreate natural protein-protein interactions. The beta-hairpin scaffold provides a versatile platform for further optimization through combinatorial substitution of the favorable mutations identified here, with the potential to reach subnanomolar affinity. Because the peptide binds independently of the GTPase nucleotide state, it could inhibit both active and inactive forms of Cdc42, a property relevant to cancers where the protein is chronically overexpressed rather than mutationally activated. These cyclized peptide inhibitors represent promising leads for therapeutic strategies targeting Cdc42-driven metastasis and resistance pathways in Ras-mutant cancers.