Cysteine Umpolung
Reflecting work in the Wang and Goldberg Groups
A longstanding challenge in peptide chemistry has been the restricted reactivity of canonical amino acids, which limits late-stage functionalization strategies. Most side chains are inherently nucleophilic, and thus nearly all peptide modification methods exploit bioconjugation—tethering electrophiles to these nucleophiles. While powerful, this paradigm leaves much of the chemical space unexplored.
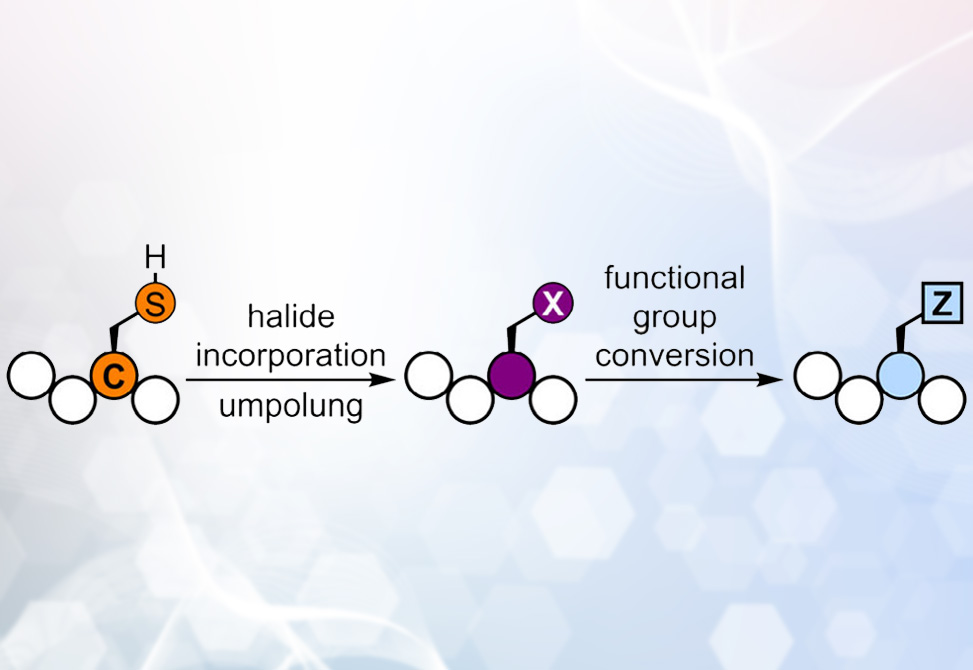
In a recent study, published in the Journal of American Chemical Society, researchers from the Wang and Goldberg Groups, at University of Rhode Island and Colgate University, respectively, introduce a fundamentally different approach: converting the highly nucleophilic cysteine thiol into an electrophilic alkyl halide, specifically Csp3–I, thereby reversing its polarity and unlocking a suite of transformations previously inaccessible in unprotected peptides.
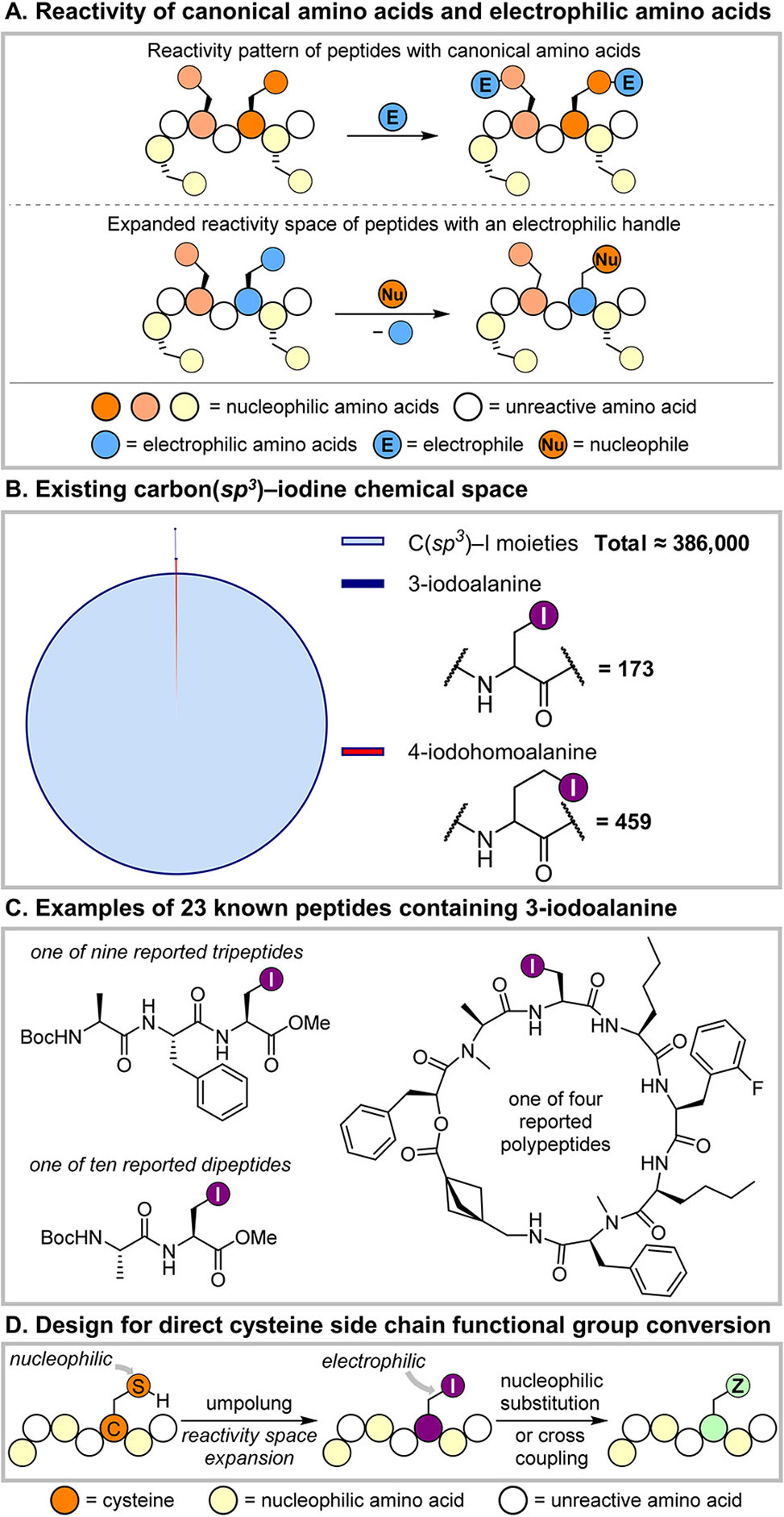
Figure 1. A| Comparison of the reactivity patterns of peptides with canonical amino acids and those with electrophilic side chains. B| The high abundance of alkyl iodides, Csp3–I, and the relative paucity of these functionalities in amino acids and peptides. C| Select examples of 23 reported peptides containing the 3-iodoalanine unit, none bearing strongly nucleophilic sites. D| Proposed strategy for incorporating an electrophilic Csp3–I moiety into peptides and potential synthetic applications.
The concept rests on a simple but daring hypothesis: that alkyl iodides, long recognized as versatile synthetic handles in small-molecule chemistry, might persist within the dense, reactive environment of peptides. Achieving this required designing a robust “one-pot” desulfoiodination strategy, wherein cysteine residues undergo a disulfide exchange, then react with electron-deficient phosphines to generate phosphonium sulfides capable of clean iodination. After careful screening, the team identified tri(3-methylthiophen-2-yl)phosphine as a key reagent, enabling nearly quantitative conversion of cysteine to 3-iodoalanine under mild, aqueous conditions.
This electrophilic editing method proved broadly applicable. Complex peptides bearing lysine, arginine, tyrosine, or aspartic acid—all potentially competing nucleophiles—were efficiently iodinated, and the resulting products showed no evidence of racemization, a persistent drawback in other cysteine-to-dehydroalanine methods. The new chemistry also tolerated proximity effects, with successful modification even when multiple reactive side chains were clustered near the cysteine target site. Stability assays revealed that the newly installed C–I bonds persisted for many hours under physiological buffer conditions, sufficient to enable downstream transformations.
Critically, the iodinated peptides could undergo a wide array of subsequent modifications. Direct thiolation converted them into lanthipeptide-like structures; selective acylation generated site-specific Nε-acetyllysine residues adjacent to cysteine; and even demanding cross-coupling reactions such as nickel-mediated Negishi arylations succeeded in the context of unprotected peptides. Together, these results show that iodinated residues function as orthogonal handles, opening a chemical toolbox typically reserved for small organic molecules but now applicable to biomacromolecules.
The implications are wide-reaching. By reengineering cysteine from a nucleophile into an electrophile, the authors demonstrate an “umpolung” strategy that bypasses the constraints of canonical amino acid chemistry. This innovation offers a new platform for late-stage peptide diversification, with potential in probing protein function, engineering biomaterials, and designing therapeutics. Beyond cysteine itself, the work suggests a broader design principle: rethinking side-chain reactivity as something to be converted rather than merely conjugated. In doing so, peptide science takes a significant step toward the vast and largely untapped chemical space of electrophilic biomolecules.
Publication Information
Author Information
![]()
Meet Dr. Daniel Honeycutt, a recent Ph.D. graduate in chemistry from the University of Rhode Island, who shifted from polymer science into peptide chemistry to tackle some of the most challenging problems in biomolecular modification.
Working in Professor Fang Wang’s laboratory, Honeycutt developed innovative methods for reengineering cysteine residues — one of peptide chemistry’s most versatile yet limiting amino acids. His research led to a rapid pyridinium-based thiol labeling strategy and a new approach for converting cysteine into β-haloalanines, creating powerful electrophilic handles for downstream transformations.
His pioneering work, recently published in the Journal of the American Chemical Society, introduces a bold “cysteine umpolung” strategy that reverses the polarity of cysteine, opening previously inaccessible chemical space for peptide diversification. Honeycutt’s research promises new opportunities in chemical biology, biomaterials, and therapeutic design.