Degrading Light
Reflecting work in the Lou Group
Targeted protein degradation has rapidly matured into a transformative drug discovery approach, offering ways to dismantle proteins once deemed undruggable. While classical proteolysis-targeting chimeras, PROTACs, exploit the ubiquitin–proteasome system to eliminate disease-relevant proteins, peptide-based degraders promise complementary advantages, such as biocompatibility and sequence programmability. Yet, conventional peptide PROTACs face two persistent limitations: they provide no real-time feedback on degradation dynamics, and they often suffer from limited pharmacokinetic stability.
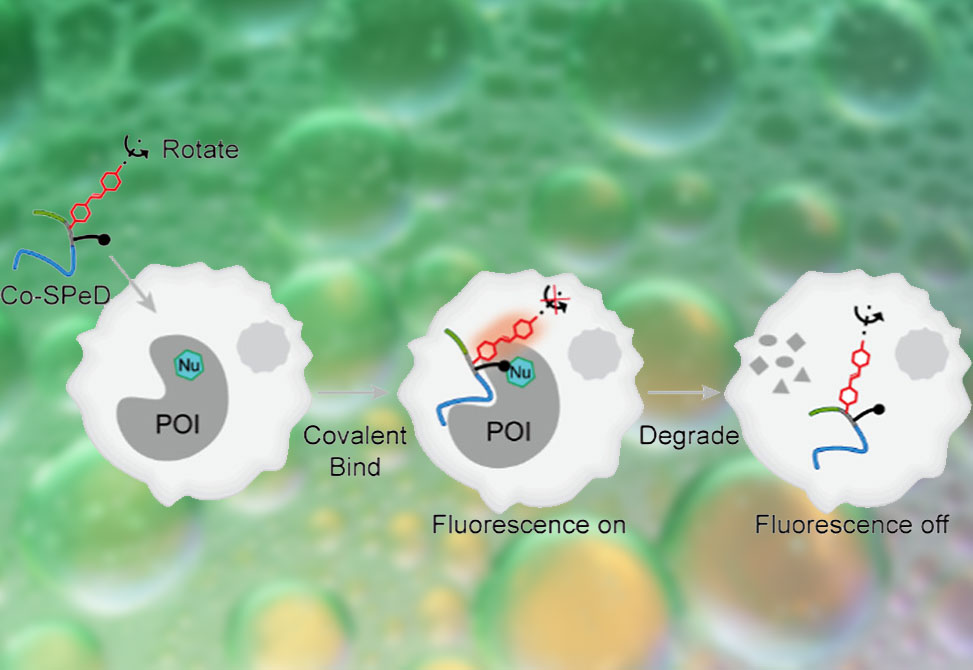
To address these challenges, researchers from the Xiaoding Lou Group at the China University of Geosciences, Wuhan, published in the Journal of the American Chemical Society, developed a covalent self-reporting peptide degrader, Co-SPeD, a modular construct designed to integrate target recognition, covalent stabilization, fluorescence reporting, and E3 ligase recruitment. The design featured four elements: 1| a peptide ligand for specific protein recognition; 2| a fluorine-substituted aryl fluorosulfate, FAFS, warhead that undergoes SuFEx chemistry with Lys, His, or Tyr residues; 3| a computer-screened rotor fluorophore, whose intramolecular rotation is restricted upon protein engagement to yield an “off–on” fluorescence signal; and 4| a peptide unit to recruit the VHL E3 ligase, thereby initiating ubiquitination and proteasomal degradation of the protein of interest, POI. The consequence is a degrader that first lights up upon binding to the POI and subsequently dims as the POI is destroyed, offering real-time fluorescence feedback on protein fate in cells and in vivo.
Design and Mechanism
Initial studies employed stilbene-derived rotor fluorophores, R1–4, whose emission properties were highly sensitive to viscosity. Docking simulations identified R2 as an optimal module for conjugation with an MDM2-binding peptide, yielding TR2. To further stabilize the probe, the FAFS warhead was incorporated, producing TFR2. Mass spectrometry, SDS-PAGE, and fluorescence gel imaging confirmed covalent bond formation between TFR2 and MDM2, specifically at the K51 residue. Compared to noncovalent TR2, TFR2 showed enhanced fluorescence response, increased anisotropy, and superior cellular labeling. Importantly, TFR2 selectively illuminated MDM2-high MCF-7 cells, produced negligible background with homologous proteins such as MDMX, and supported imaging of tumor xenografts in vivo.
Adding a VHL-binding motif to TFR2 created TFR2-P, the full Co-SPeD prototype. In living cells, fluorescence initially increased as TFR2-P engaged MDM2, then declined in parallel with protein degradation. Western blotting confirmed MDM2 depletion and concomitant stabilization of p53. Fluorescence loss was blocked by proteasome inhibition or VHL knockdown, consistent with UPS-mediated degradation. TFR2-P displayed selective cytotoxicity toward MDM2-high tumor cells and suppressed their proliferation. In xenograft models, subcutaneous dosing of TFR2-P reduced tumor burden by 76% without systemic toxicity.
Generality Across Targets
Beyond MDM2, the Co-SPeD strategy was extended to additional proteins, including BCL-xL, GRP78, and the “undruggable” KRAS(G12D). For each, peptide recognition motifs were combined with distinct rotor fluorophores and the FAFS warhead to create degraders capable of both imaging and depletion. In HeLa, HepG2, and A427 cells, these constructs exhibited concentration-dependent fluorescence decline matched to target knockdown, as well as functional inhibition of downstream signaling such as AKT/ERK phosphorylation in KRAS(G12D) cells. These results underscore the versatility of Co-SPeDs across diverse protein classes, including challenging oncogenic drivers.
Therapeutic Implications
Real-time visualization of protein degradation was leveraged to optimize combination therapy with cisplatin. In cell culture, cisplatin proved most effective when administered at time points corresponding to minimal MDM2 fluorescence, in essence, after significant target depletion by TFR2-P. Translating this to xenograft models, cisplatin dosing guided by TFR2-P fluorescence monitoring suppressed tumor growth by 95% compared with only 45% inhibition in unguided regimens. These studies demonstrate the potential of Co-SPeDs not only as degraders but as dynamic reporters that can temporally optimize therapeutic interventions.
Conclusions
The Co-SPeD platform represents a new generation of peptide-based PROTACs that unify target degradation with built-in fluorescence reporting. By exploiting covalent SuFEx chemistry and rotor-fluorophore signaling, these molecules achieve durable protein engagement, selective ubiquitin–proteasome degradation, and live feedback on degradation kinetics. The ability to both destroy and visualize proteins in real time enables rational tuning of combination therapies, as shown with cisplatin, and extends targeted degradation to difficult cancer drivers such as KRAS(G12D). This dual-function design positions Co-SPeDs as powerful tools for chemical biology and therapeutic innovation in precision oncology.