Detection of Pulmonary Fibrosis
Reflecting work in the Raines
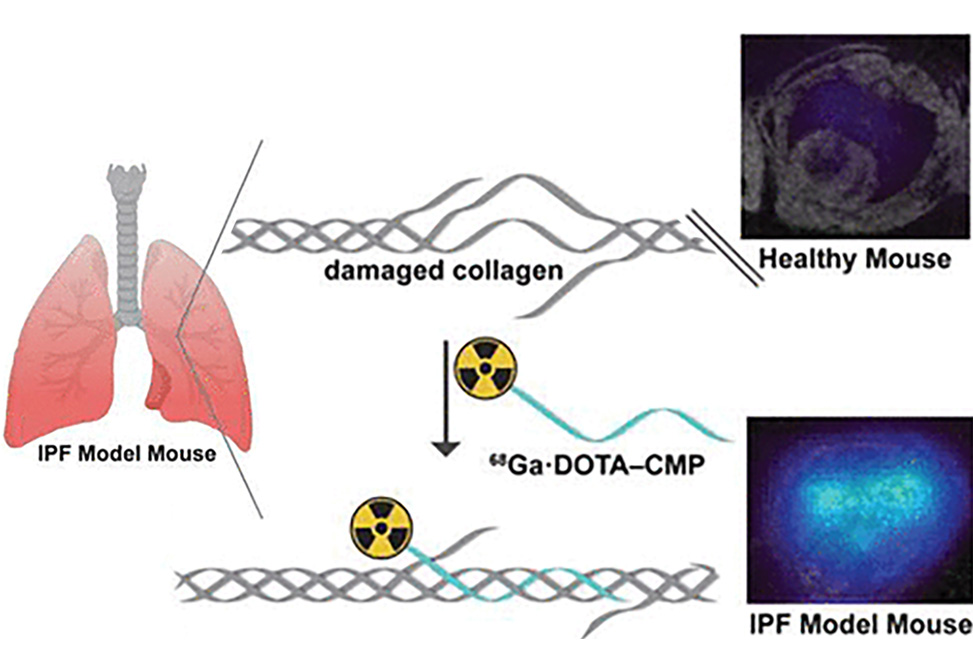
Idiopathic pulmonary fibrosis, IPF, is a disease of unknown etiology that is characterized by excessive deposition and abnormal remodeling of collagen. IPF has a mean survival time of only 2−5 years from diagnosis, creating a need to detect IPF at an earlier stage when treatments might be more effective. We sought to develop a minimally invasive probe that could detect molecular changes in IPF-associated collagen.
In work published in ACS Sensors, researchers in the Raines Group describe the design, synthesis, and performance of [68Ga]Ga·DOTA−CMP, which comprises a positron-emitting radioisotope linked to a collagen-mimetic peptide, CMP. This peptide mimics the natural structure of collagen and detects irregular collagen matrices by annealing to damaged collagen triple helices. Group members assessed the ability of the peptide to detect aberrant lung collagen selectively in a bleomycin-induced mouse model of pulmonary fibrosis using positron emission tomography, PET. [68Ga]Ga·DOTA−CMP PET demonstrated higher and selective uptake in a fibrotic mouse lung compared to controls, minimal background signal in adjacent organs, and rapid clearance via the renal system.
These studies suggest that [68Ga]Ga·DOTA−CMP identifies fibrotic lungs and could be useful in the early diagnosis of IPF.