Docking Bias
Reflecting work in the Keating Group
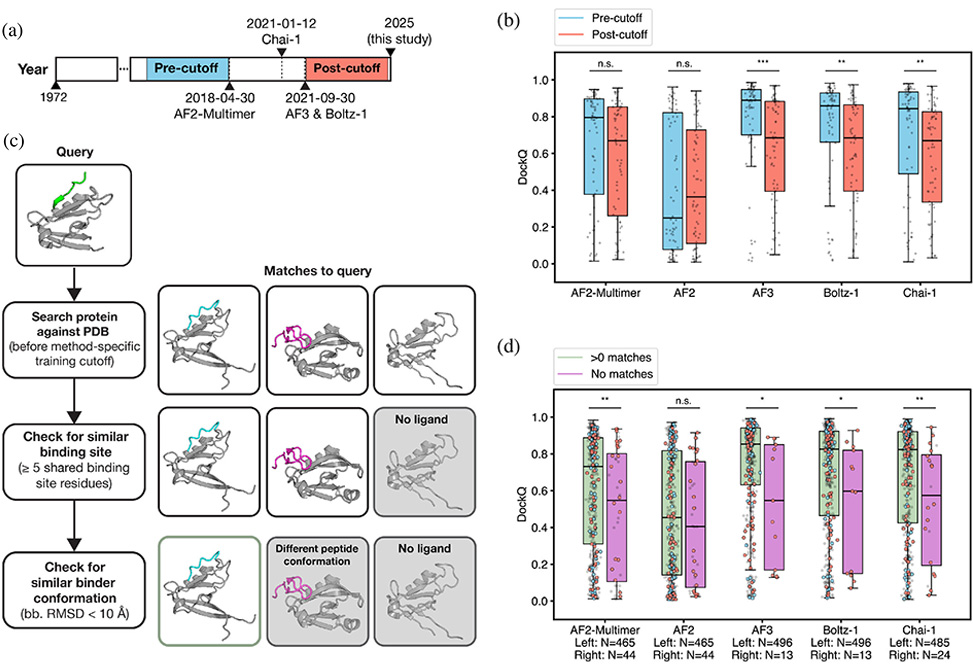
Deep-learning structure predictors now place many peptides correctly onto their protein partners, and this recent work from Lindsey Guan and Amy Keating at the Massachusetts Institute of Technology, published in Protein Science, explains why they succeed and where they fail. Benchmarking AF2, AF2-Multimer, AF3, Boltz-1, and Chai-1 on a curated, non-redundant set of 509 protein–peptide complexes confirms strong top-line accuracy—many high-quality DockQ > 0.80 predictions—while exposing two central dependencies.
Predicted confidence, ipTM+pTM, tracks quality well, Pearson > 0.7, and most high-confidence models are at least “acceptable,” DockQ > 0.23. Nevertheless, all methods still produce confident false positives—poses in the wrong pocket or with flipped peptide orientation—and AF3’s edge in atomically precise cases diminishes on truly novel targets.
Training overlap matters. Splitting targets by training-data cutoffs reveals a marked accuracy drop for AF3, Boltz-1, and Chai-1 on post-cutoff structures, whereas AF2/AF2-Multimer are less sensitive to time splits. A stricter screen that searches the training set for binding-site matches shows the same effect: interfaces represented in training are predicted substantially better. In several illustrative failures, models prefer a frequently observed “decoy” pocket from the corpus over the experimentally correct site—homology-like bias masquerading as generalization.
Contrary to common assumptions, inter-chain coevolution from paired protein–peptide MSAs contributes surprisingly little. Even when deeper paired MSAs are constructed by extending peptide context and then trimming back to the PDB-defined segment, shuffling the pairings barely perturbs accuracy for protein–peptide docking. For classic protein–protein benchmarks, shuffling can hurt modestly for AF2-Multimer and Boltz-1, but the effect is case-dependent. Interestingly, masking inter-chain attention in AF2-Multimer’s Evoformer degrades performance on a subset of cases, implying that the network can implicitly extract inter-chain signals even from unpaired MSAs via the interleaved row/column attention.
What does matter is the receptor’s unpaired MSA. The protein’s evolutionary profile drives binding-site selection, a result made vivid by ablations that remove peptide information entirely: masking the peptide as “X” leaves 40–51% of previously successful cases still successful, meaning the model can identify the pocket and approximate the bound pose without residue-level cues from the ligand. Swapping MSAs for structural templates further shows that AF2-Multimer loses more when deprived of the protein MSA than when deprived of peptide evolution, indicating that Evoformer’s processing of the receptor MSA encodes information templates alone do not replace.
When peptide MSAs are reasonably deep, ≥50 sequences, removing them produces a modest but consistent drop in DockQ for about one-fifth of complexes, with errors in both pocket choice and local peptide geometry. Structural templates for either partner can partially rescue performance, and AF3 in particular makes strong use of good templates when MSAs are absent. For AF2-Multimer, however, replacing the receptor MSA with a structural template is more damaging than losing the template itself, indicating that the Evoformer extracts binding-site information from the protein MSA that templates cannot substitute.
The broader message for peptide science and design is pragmatic. Date-based splits alone overestimate generalization; binding-site similarity filters are needed to gauge performance on truly novel interfaces. Ensuring that a high-quality target MSA or an appropriate structural template is available is essential. Minimal experimental priors—a pocket triad or a single restraint—can sharpen poses, but high model confidence does not guarantee correctness when targeting new sites or under-represented folds. Training-aware benchmarks reveal when a prediction is genuinely beyond the models’ comfort zone.
Publication Information
Author Information

Lindsey Guan is a Ph.D. candidate in the Computational and Systems Biology program at MIT, working in the lab of Professor Amy Keating. Lindsey’s research focuses on building computational methods for the modeling and design of peptide or mini-protein binders. In particular, she is interested in methods for designing highly target-discriminative binders. Previously, she earned a dual Bachelor’s degrees in Computer Science and Microbial Biology at the University of California, Berkeley, and worked on structural bioinformatics in the lab of Professor Steven Brenner.