The total synthesis and process development of enlicitide decanoate, MK-0616, an orally bioavailable inhibitor of proprotein convertase subtilisin/kexin type 9, PCSK9, represents a major breakthrough in macrocyclic peptide synthesis. This innovative molecule is under development as a therapeutic for atherosclerotic cardiovascular disease, ASCVD, addressing a critical need for effective and patient-friendly cholesterol management therapies. Unlike previous PCSK9 inhibitors, which are monoclonal antibodies requiring parenteral administration, enlicitide is an entirely synthetic small molecule that can be taken orally. This distinction necessitated the development of a scalable, cost-effective, and efficient synthetic strategy capable of meeting anticipated global demand.
The structural complexity of enlicitide posed a significant challenge in synthetic chemistry. The molecule comprises eight amino acid residues, six of which are noncanonical, alongside two macrocyclic domains, including a 37-membered macrocycle incorporating an elaborated non-peptidic fragment. Additionally, an ammonium-containing side chain critical for biological activity further complicated synthesis, given its polarity and impact on solubility. These structural features ruled out standard solid-phase peptide synthesis, SPPS, demanding a fully modular and convergent approach that would allow for industrial-scale production.
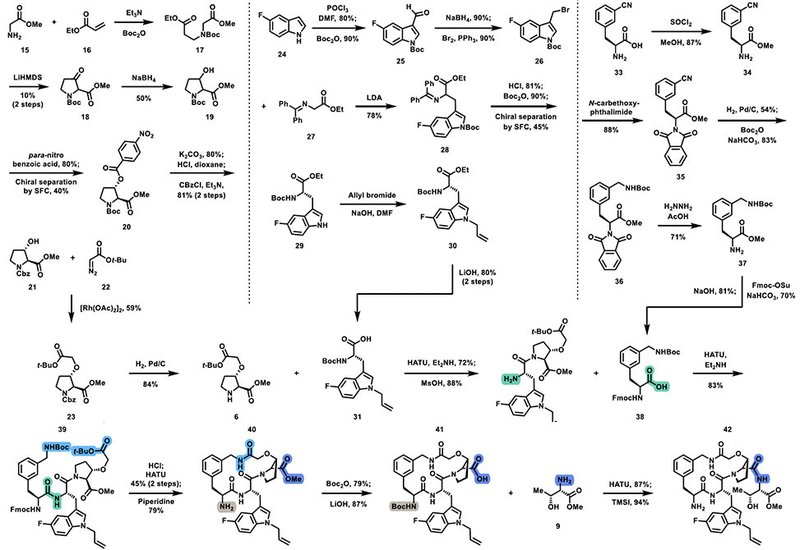
The first-generation synthesis of enlicitide, though a valuable proof of concept, was ultimately deemed impractical for commercial manufacturing. Spanning 63 total steps with 28 in the longest linear sequence, the process relied heavily on chromatography for purification and suffered from significant yield inefficiencies. The overall yield was so low that producing even a single 100 kg batch would require processing over 170 metric tons of early-stage intermediates. The synthetic strategy employed ring-closing metathesis, RCM, to construct the macrocyclic core and reserved the amidation of the ammonium side chain for the final steps. Despite successfully assembling the target molecule, the first-generation synthesis could not support large-scale production due to its inefficiencies.
Scheme 8. Second Generation End Game Synthesis of Enlicitide
Recognizing these limitations, researchers at Merck developed a second-generation synthesis, significantly improving process efficiency, cost, and scalability. A key innovation in this revised approach was the strategic restructuring of the synthetic sequence to delay macrocyclization, thereby enhancing yield and minimizing chromatographic purification. The discovery of three crystalline intermediates enabled purification through crystallization rather than chromatography, streamlining the overall process. By refining the assembly of the Northern and Southern fragments, the Merck team achieved a dramatic reduction in step count—bringing the total down to 43 steps, with the longest linear sequence reduced to 21. Yield improvements were equally remarkable, increasing by a factor of 1000 compared to the first-generation synthesis. The process also incorporated a biocatalytic and chemoenzymatic strategy that simplified the synthesis of key noncanonical amino acids, enhancing both throughput and stereochemical control.
Published in the Journal of the American Chemical Society, this work underscores the clinical relevance of enlicitide decanoate. PCSK9 inhibitors have emerged as a transformative class of lipid-lowering therapies, reducing low-density lipoprotein cholesterol, LDL-C, by enhancing LDL receptor recycling. Until now, approved PCSK9 inhibitors, such as evolocumab and alirocumab, have been limited to injectable formulations, presenting barriers to widespread adoption. Enlicitide decanoate, as the first oral PCSK9 inhibitor, represents a paradigm shift in the field. Its high oral bioavailability, coupled with a convenient daily dosing regimen, addresses the major limitations of current therapies. The molecule has already demonstrated significant LDL-C reduction and excellent tolerability in Phase 2 clinical trials, positioning it as a highly promising candidate for global cholesterol management.
Beyond its immediate therapeutic implications, the successful synthesis of enlicitide marks a major advancement in macrocyclic peptide drug development. Historically, large-scale production of peptides has relied on fermentation or recombinant expression, with chemical synthesis largely reserved for smaller, less complex molecules. The development of a fully synthetic, industrially scalable route to enlicitide establishes a precedent for future macrocyclic peptide drugs, expanding the possibilities for chemically manufactured peptide therapeutics. The lessons learned from this endeavor—particularly in designing modular synthetic strategies, leveraging crystallization-driven purification, and optimizing macrocyclization sequences—will undoubtedly inform the development of next-generation bioactive peptides.
The Merck team concludes by emphasizing the broader impact of their work, not only in accelerating enlicitide’s clinical progress but also in setting a new benchmark for macrocyclic drug synthesis. By successfully bridging the gap between synthetic peptide chemistry and large-scale manufacturability, they have laid the foundation for a future in which complex bioactive peptides can be efficiently produced at commercial volumes. This work represents a compelling demonstration of how cutting-edge synthetic chemistry can drive innovation in pharmaceutical development, ultimately improving access to life-saving therapeutics worldwide.