Iodo Quantification
Reflecting work in the Lilienkampf Lab
In work published in ACS Chemical Biology, David J. Clarke, Annamaria Lilienkampf, and collaborators from the University of Edinburgh, introduce an iodo based labeling strategy that converts matrix assisted laser desorption ionization mass spectrometry into a reliable quantitative tool for screening peptides and peptide libraries. Conventional MALDI MS suffers from heterogeneous matrix crystallization, ionization suppression, and narrow dynamic range, which makes accurate quantification difficult. Existing approaches often rely on isotope labeling with deuterium, carbon thirteen, or nitrogen fifteen or on subtle structural modifications such as methylation or glycine deletion. These methods can be costly or sequence dependent, and the resulting analyte and internal standard peaks frequently lie too close in mass for confident resolution in mixtures of related sequences. Specialized ionic liquid matrices improve spot homogeneity but require extensive optimization for each sample type and remain poorly suited for high throughput workflows. MALDI MS offers clear advantages in speed, cost, and parallelization, especially in multisample plate formats, yet its lack of quantification has limited its use in peptide library screening.
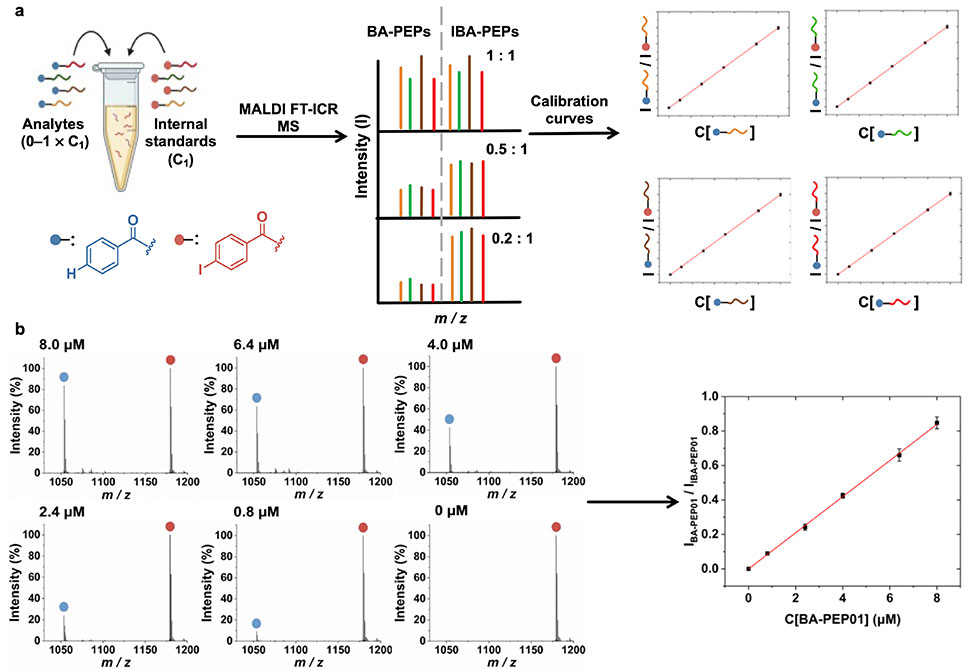
The authors address this limitation by tagging peptides at the N terminus with either benzoic acid or 4 iodobenozoic acid to generate pairs that differ only by a single iodine atom. Iodine provides an attractive mass defect because it exists as a single stable isotope and introduces a large negative mass offset that cleanly separates analyte and internal standard peaks in high resolution MALDI FT ICR MS. The tagging procedure is synthetically straightforward, requiring only one additional coupling step during standard Fmoc solid phase peptide synthesis. Initial validation used four short peptides designed to probe charge, hydrophobicity, and sequence variation. Calibration curves across multiple concentration ranges for each BA and IBA pair showed excellent linearity with coefficients of variation below five percent, and the method remained robust down to low micromolar concentrations. The modified matrix one methylimidazole coupled α cyano four hydroxycinnamic acid yielded uniform crystallization and reproducible ionization, outperforming conventional CHCA in spot consistency and signal stability.
The strategy was then evaluated with mixtures of four peptides. BA tagged analytes and IBA tagged internal standards were combined in defined ratios and analyzed by MALDI FT ICR MS. Each analyte to internal standard peak ratio tracked linearly with analyte concentration, and independent mixture sets with distinct concentration profiles were quantified with errors below five percent. These results demonstrate that the IBA approach supports accurate quantification in mixed peptide samples and preserves analyte integrity across varied sequences. To test performance in a biological context, the authors incubated peptides with fixed Escherichia coli and Bacillus subtilis, collected the supernatants, and quantified residual peptide concentrations. Analytes with higher net positive charge, such as BA PEP01, showed strong binding to both bacteria, while neutral peptides such as BA PEP04 showed no detectable binding. Fluorescence imaging using FAM tagged analogs confirmed binding trends and validated MALDI based quantification.
Building on this foundation, the authors synthesized a twelve member glycine zipper peptide library using a KGLTAGLTAGL template varied at two positions. All twenty four signals corresponding to BA and IBA tagged versions appeared as distinct, resolvable peaks in the MALDI FT ICR MS spectrum. A notable example involved the M plus four isotopologue of the largest BA tagged peptide lying only 0.16 daltons from the monoisotopic peak of the smallest IBA tagged peptide, yet the iodine mass defect ensured complete separation. Calibration curves for all twelve peptides remained linear with high reproducibility. Screening the library against E. coli and B. subtilis revealed clear binding preferences. Peptides with lysine or phenylalanine at variable positions showed strong binding, while threonine or serine reduced binding. BA FK, BA LK, and BA TK exhibited complete binding to B. subtilis and more than fifty percent binding to E. coli. Fluorescence microscopy of FAM tagged peptides corroborated MALDI derived binding patterns and confirmed that sequence dependent physicochemical features, rather than antimicrobial potency, drive bacterial association in this family.
The authors then extended the approach to a one hundred twenty five member library incorporating noncanonical amino acids. Expansion of the glycine zipper motif allowed exploration of hydrophobic, hydrophilic, and charged residues at multiple positions. Ninety nine BA and IBA pairs were detected in the MALDI FT ICR MS spectrum. Missing sequences were attributed to ionization suppression by highly charged members because undetected peptides carried net charges of minus one or zero, with only a few at plus one. Even with this spectral complexity, each resolvable BA and IBA peak pair maintained a linear peak ratio to concentration relationship. Screening against E. coli and B. subtilis produced a distribution of binding percentages from zero to sixty, and sequence analysis highlighted the importance of hydrophobic and positively charged residues in enhancing binding. A small group of peptides showed species specific preferences, such as BA Knb binding B. subtilis preferentially and BA Ksb showing improved binding to E. coli. Resynthesized peptides tagged with NBD or FAM confirmed selective labeling patterns, with NBD providing sharper discrimination due to its environmental sensitivity and minimal steric influence. Importantly, these high binders lacked strong antimicrobial activity, with minimum inhibitory concentrations above fifty micromolar, which underscores the distinction between binding affinity and bactericidal function. Circular dichroism spectroscopy revealed minimal secondary structure in aqueous solution, implying that binding is driven by primary sequence properties rather than ordered amphipathic helices or β sheets.
Overall, this iodo based mass defect labeling strategy provides a practical and broadly applicable solution for quantitative MALDI FT ICR MS analysis of peptide libraries. The single iodine modification creates a stable internal standard with clear mass separation, enabling accurate quantification of individual peptides, mixtures, and complex libraries. The method integrates seamlessly into standard solid phase synthesis, requires no isotopic reagents, and supports parallel analysis in multiwell formats, which expands throughput while maintaining measurement precision. By enabling quantitative assessment of peptide binding to bacterial cells in solution, this approach accelerates discovery of bacteria binding peptides and offers a versatile platform for applications in diagnostics, imaging, and peptide based probe development.
Publication Information
Author Information

Liao Hu earned a BEng in Pharmaceutical Engineering from the East China University of Technology before moving to the UK to complete an MSc in Medicinal and Biological Chemistry at the University of Edinburgh in 2020. He completed his Ph.D. in 2025 in the Lilienkampf group at the School of Chemistry, University of Edinburgh, where he developed high-throughput screening platforms for bacteria-binding peptides. Liao is now a postdoctoral researcher in the group of Rebecca Beveridge at the University of Strathclyde, focusing on protein mass spectrometry to investigate S-acylation.