Lanthipeptide Modeling
Reflecting work in the Walker Lab
Lanthipeptides, the thioether-crosslinked members of the RiPP family, are compelling antibiotic scaffolds. Yet, they have remained largely outside the reach of structure-based design. Standard modeling tools, for example, Rosetta, do not natively understand lanthionine/methyllanthionine rings or dehydrated residues, dehydroalanine, dehydrobutyrine, blocking predictive modeling and rational engineering. Researchers in the Walker and Meiler Groups at Vanderbilt University, published in Protein Science, remove that roadblock by parameterizing these chemistries in Rosetta and demonstrating that ensemble-based refinement against NMR data captures the intrinsic flexibility of lanthipeptides while maintaining experimental agreement and improving energetic quality. The result is a practical platform for lanthipeptide prediction and design.
Two capabilities were added to Rosetta. First, bonded parameters for thioether crosslinks were introduced: C–S bond distance, two adjacent angles, and three dihedrals for lanthionine; for methyllanthionine, additional angle terms and an improper dihedral maintain methyl geometry. Torsional terms draw from Amber protein side-chain potentials, with bond/angle terms using Rosetta’s Cartesian energy. Second, backbone conformational statistics for dehydrated residues were calculated by DFT-based φ/ψ scans, (M05-2X/6-311G(d,p)), yielding new Ramachandran tables that reflect the sp2 α-carbon and, for dehydrobutyrine, restricted regions to avoid γ-sterics.
Structure prediction then proceeds by generating conformational ensembles that are scored by Rosetta energy and fit to NMR NOE restraints in an ensemble-averaged sense, in essence, the ensemble collectively satisfies distances, no single member must meet all. This is crucial: enforcing per-structure satisfaction often optimizes towards a single conformation, averaging exposes multiple low-energy states that are altogether consistent with experimental data.
Across ten PDB entries, helical and non-helical, Rosetta ensembles refined with NOEs show:
Similar agreement to experimental NOE data compared to deposited NMR ensembles,
Lower Rosetta energies, often improved fa_atr, fa_rep, fa_sol, fa_elec, rama_prepro, and
Greater conformational breadth, higher intra-ensemble backbone RMSD.
As a control, selecting 20 low-energy, NOE-consistent Rosetta structures without explicit ensemble-averaging still improved energies versus PDB, but full ensemble averaging produced the lowest energies and broadest yet data-consistent flexibility. These trends suggest the deposited ensembles may underestimate true flexibility, and that the Rosetta ensembles map lower free-energy basins compatible with NMR data.
Helical lanthipeptides benefit from AlphaFold2 as a starting backbone, for helicity only, after which lanthionine formation, dehydration, and conformational sampling in Rosetta refine the models. A consistent pattern emerges:
Rings embedded in helices are locally rigid, ring RMSD typically ≤1 Å vs PDB,
Global variability concentrates in tails and inter-helix linkers, frequently enriched in glycine.
Representative cases highlight this division of labor: glycine-rich segments, for example, GLGV motifs, serve as hinges; adjacent rings at termini perturb helical packing; and ring-mediated contacts modulate inter-helix orientation. Notably, allowing two alternative nonhelical “bites” in a glycine-rich stretch can reconcile NOE contacts with helical chemical-shift predictions, underscoring the value of ensembles for resolving apparently conflicting data modalities.
Non-helical lanthipeptides require broader sampling: randomized φ/ψ guided by the Ramachandran potential,iterative ring closure, and relaxation. Here, ring–ring orientation and uncyclized segments dominate flexibility. Even when individual rings are moderately rigid, relative ring orientations vary by 2–8 Å backbone RMSD vs PDB. Where rings lack internal hydrogen bonds and include glycine, Rosetta finds multiple low-energy ring conformers, with the largest, H-bond-poor, Gly-rich macrocycles showing the greatest ring RMSD. Conversely, regions densely cyclized, overlapping/nested rings, are locally more rigid, yet global rigidity is not guaranteed: cyclization reduces but does not eliminate conformational freedom.
For peptides with overlapping rings, Rosetta sampling uncovers non-interconverting atropisomers, kinetically trapped arrangements. Pre-selection filters these when they conflict with experimental restraints; the PDB-major isomer typically dominates the candidate pool. Recognizing atropisomerism is essential when interpreting NMR for nested/overlapping macrocycles.
Three design-relevant determinants recur:
Uncyclized regions, termini, inter-ring linkers, are principal sources of global motion.
Ring microchemistry matters: internal H-bond networks and β-branched/proline residues stabilize; glycine and H-bond paucity promote mobility.
Topology, adjacent/overlapping/nested, tunes local rigidity but does not alone lock the global fold.
Two bottlenecks remain for truly de novo prediction: 1| current pipelines assume known ring topology and stereochemistry; these depend on enzyme–substrate pairing and are not yet predictable from sequence alone, and 2| widely used chemical-shift tools, for example, TALOS-N, SHIFTX2, do not handle noncanonical residues, limiting orthogonal restraints. Extending shift predictors to Dha/Dhb/lanthionine, and their neighbors, would sharpen ensemble selection. Finally, AlphaFold’s helicity cues are helpful but not decisive; ring patterns incompatible with helices should route models to the non-helical pipeline.
With lanthionine and dehydration chemistries now native to Rosetta, lanthipeptide design can move beyond brute-force library screens. The demonstrated ensemble-centric refinement provides realistic, low-energy structural hypotheses that 1| explain disparate NMR observables, 2| reveal motion hotspots for engineering, for example, rigidify a linker, introduce an intra-ring H-bond, replace Gly, and 3| guide screen construction or hit optimization for antibiotic and targeting applications.
Encoding lanthionine crosslinks and dehydrated residues in Rosetta, combined with ensemble-averaged NMR restraint fitting, yields experiment-consistent, lower-energy, and more flexible models than deposited NMR ensembles. The picture that emerges is of dynamically adaptive macrocycles: rings can enforce local order, but global behavior is set by where and how those rings are placed, what residues they contain, and how they interact. This capability turns lanthipeptides into first-class citizens of structure-based design, enabling hypothesis-driven engineering of macrocyclic antibiotics and beyond.
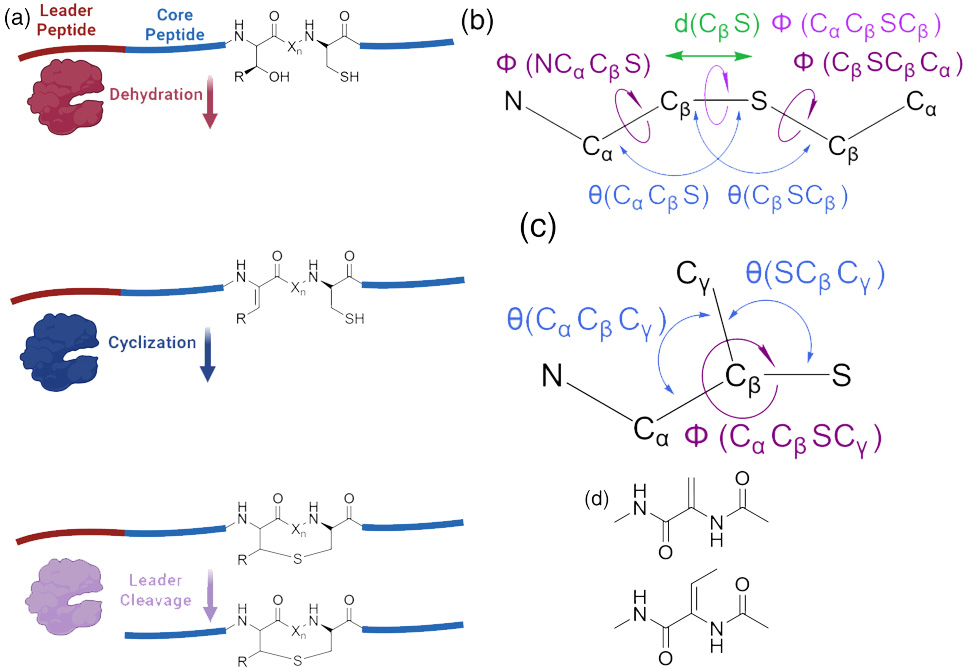
Lanthipeptides are ribosomally synthesized and post-translationally modified peptides. a| Lanthipeptides contain a leader peptide for enzymatic recognition and a core peptide that is modified. Serine and threonine residues are dehydrated in lanthipeptides and serve as Michael acceptors for nucleophilic attack by cysteine residues to form lanthionine rings. b| For lanthionine rings, the important parameters for modeling the cyclization are the new bond distance and the two angles and three dihedrals involving this bond. c| For methyllanthionine rings, the addition of bond angle terms and an improper dihedral term involving the methyl groups helps to maintain the proper geometry. d| The dehydrated amino acids are often seen in the final lanthipeptide product and are important to model. Shown here are dehydroalanine, top and dehydrobutyrine, bottom. Created in BioRender. Tydings, C., 2025
Author Information

Meet Claiborne Tydings, a fifth-year Ph.D. candidate in chemistry at Vanderbilt University. He develops and applies tools for computational peptide modeling to design therapeutic peptides. These tools include implementing and benchmarking lanthipeptide modeling in the Rosetta macromolecular modeling software.
His innovative work has earned recognition through NIH T32 and F31 awards that focus on developing peptide therapeutics for obesity and cancer. Outside of the lab, Claiborne is an avid distance runner and has qualified for the Boston Marathon.