Macrocyclic Peptide Antibiotics
Reflecting recent work in the van der Donk Group
Ribosomally synthesized and posttranslationally modified peptides, RiPPs, represent one of the most structurally diverse classes of natural products, yet many RiPP families remain biosynthetically uncharacterized. In a landmark study, published in the Journal of the American Chemical Society, the van der Donk group at the University of Illinois at Urbana, elucidates the complete biosynthetic pathway for a macrocyclic peptide antibiotic that bears close structural resemblance to the historically enigmatic biphenomycins. This discovery not only solves a decades-old question about the biosynthetic origin of these antibiotics but also expands the functional repertoire of several important enzyme families.

The van der Donk Group
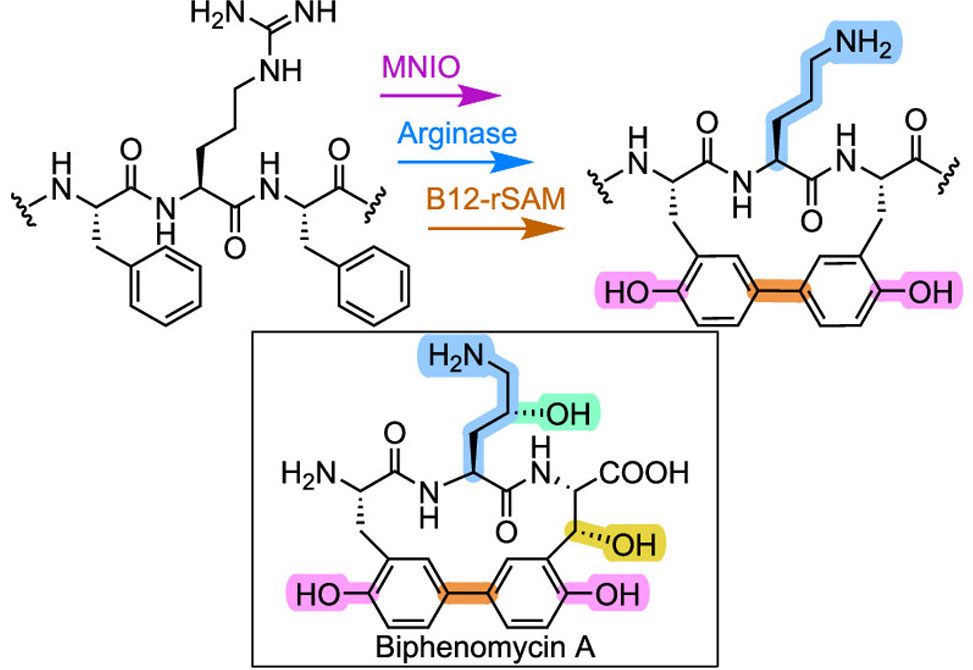
The team, which also includes the Challis and Cryle groups at Monash University in Australia, identified a conserved FHAFRF motif within a group of RiPP precursor peptides encoded biosynthetic gene clusters, BGCs, that encode in multinuclear nonheme iron-dependent oxidative enzymes, MNIOs. Focusing on one such BGC from Peribacillus simplex, designated the pbs cluster, they reconstructed the pathway in E. coli and characterized the activities of five distinct enzymes: a MNIO – PbsC, a RiPP recognition element-containing partner protein – PbsD, an arginase – PbsE, a cupin-like hydroxylase – PbsQ, and a B12-dependent radical SAM enzyme – PbsB.
This biosynthetic sequence proceeds in a highly ordered fashion. PbsC and PbsD catalyze the regioselective ortho-hydroxylation of two phenylalanine residues within the FRF motif, yielding ortho-tyrosines. PbsE then converts the adjacent arginine into ornithine, but only when the substrate has been doubly hydroxylated – highlighting the strict substrate selectivity. PbsQ further hydroxylates the newly formed ornithine, and PbsB catalyzes a C–C cross-link between the aromatic rings of the ortho-tyrosines, forming a rigid biaryl macrocycle. The final maturation step, cleavage of the leader peptide, is carried out by a metalloprotease, PbsP.
Using tandem mass spectrometry and multidimensional NMR, the team confirmed the precise site and order of each transformation. Notably, the cross-linking reaction catalyzed by the B12-rSAM enzyme PbsB represents the first such example of C–C bond formation between aromatic residues by this enzyme class. The MNIO-mediated ortho-hydroxylation of phenylalanine residues is also unprecedented and marks a significant expansion of known MNIO function, which had previously been limited to Cys, Asn, and Asp residues.
The mature macrocyclic product closely mimics the structure of biphenomycin antibiotics, which were first isolated in the 1960s but whose biosynthetic origins remained undefined. Sequencing the genome of a known biphenomycin producer, Streptomyces filipinensis, revealed a highly homologous BGC, bpm cluster, strongly supporting a shared biosynthetic logic.
The researchers in the van der Donk team further employed AlphaFold3 to model protein–peptide interactions. The predicted structures revealed canonical β-sheet interactions between the precursor leader peptides and the RRE domain of PbsD and placed catalytically important residues in spatial proximity to active site metals, consistent with experimental data.
This work offers a compelling example of biosynthetic logic that combines strict substrate ordering with broad chemical novelty. It establishes a clear enzymatic route to biphenomycin-like compounds, offers a framework for analog production, and demonstrates the utility of combining genome mining, enzymology, and structural biology. Moreover, it deorphanizes an entire class of peptide antibiotics and introduces new enzymatic tools for peptide macrocyclization and posttranslational modification.