Mirror-Image Hunters
Reflecting work in the Dijkgraf Lab
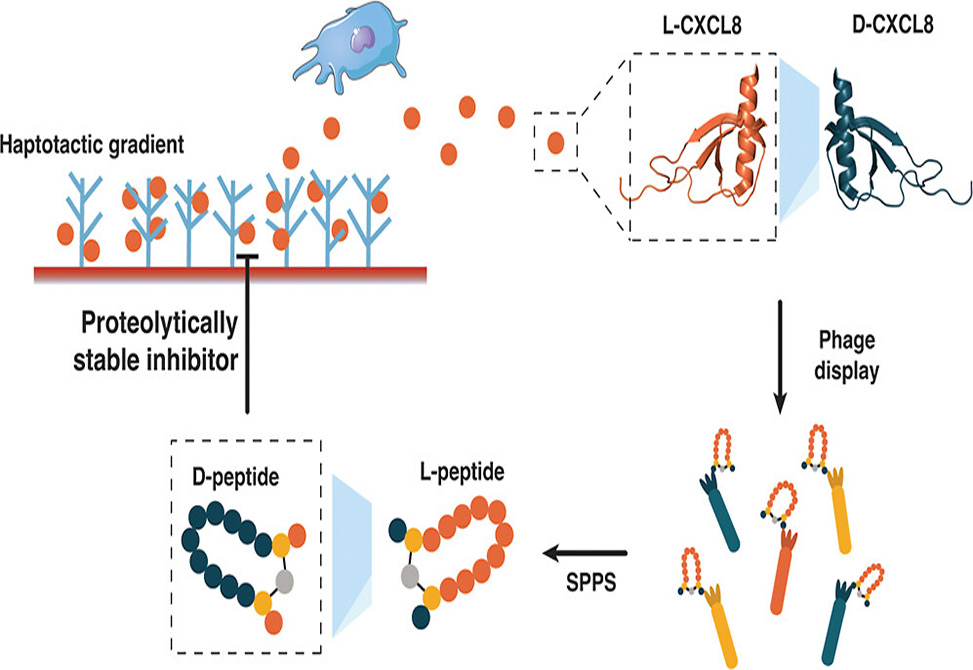
Chemokines make frustrating drug targets. These small signaling proteins orchestrate immune cell migration during inflammation, making them attractive therapeutic targets for conditions ranging from atherosclerosis to arthritis. Yet their relatively smooth surfaces lack the deep pockets where small molecules typically bind. Peptides offer a solution with their larger binding footprints, but natural L-peptides face a fundamental problem: proteases chew them up within minutes in the bloodstream. A team led by Stepan Denisov at the University of Vienna and Ingrid Dijkgraaf from Maastricht University, published in ACS Chemical Biology, has now used mirror-image phage display to identify D-peptide binders against the chemokine CXCL8, producing candidates that resist enzymatic degradation while maintaining submicromolar affinity.
The mirror-image approach exploits a clever symmetry trick. Researchers first synthesize the target protein entirely from D-amino acids, creating a mirror-image version of the natural L-protein. They then screen billions of natural L-peptide sequences against this D-target using standard phage display. Any L-peptide that binds the D-target will have a D-peptide counterpart that binds the natural L-target, because the molecular handshake works in both directions. The team synthesized D-CXCL8 through native chemical ligation of three fragments, confirmed proper folding by circular dichroism spectroscopy showing the expected mirror-image spectrum, and immobilized the synthetic chemokine on streptavidin beads for selection.
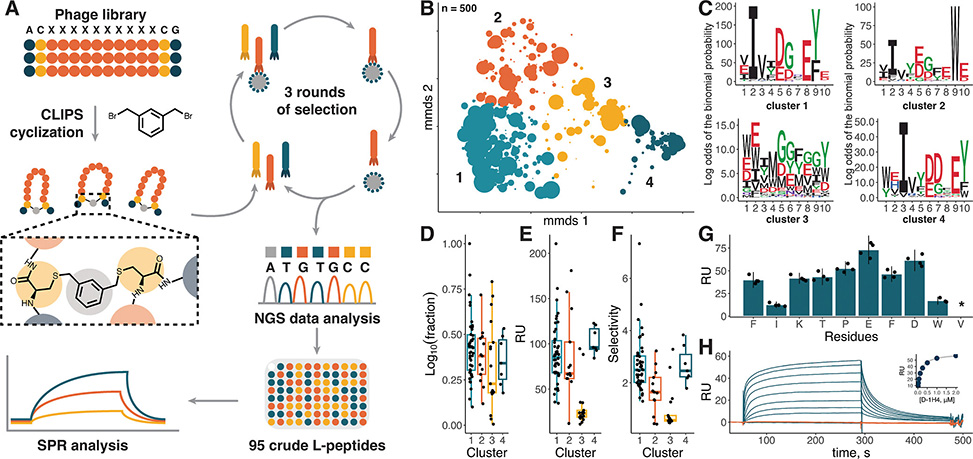
Figure 2 (A) Scheme illustrating the phage display selection pipeline. (B) 2D projection of the chemical space of 500 of the most abundant sequences after phage display selection based on sequence dissimilarity. The markers’ size is scaled proportionally to log10 of abundance in the NGS pool, clusters are shown in color and numbered. (C) pLogo plots for four clusters indicated in (B). Abundance (D), binding level to D-CXCL8 (E), and selectivity (F) of crude L-peptides selected for screening. Abundance is expressed as log10 of peptides’ fraction in the NGS pool, selectivity as a ratio of binding levels to D- and L-CXCL8. (G) Binding level of unpurified Ala mutants of L-1H4 peptide to D-CXCL8. (H) Interaction sensograms of D-1H4 with L-CXCL8 (blue) and D-CXCL8 (orange) at concentrations ranging from 15 nM to 2 μM. The binding curve based on equilibrium values is shown in the inset.
To constrain their peptide library into defined shapes, the researchers incorporated the CLIPS cyclization strategy. Two cysteine residues flanking a randomized ten-residue region react with a bis-bromomethylbenzene scaffold, locking each peptide into a cyclic conformation. Three rounds of selection against D-CXCL8 yielded roughly 2000-fold enrichment. Next-generation sequencing of the selected pool revealed four distinct sequence clusters, three of which shared variations of an Ile/Leu-Asp-Gly-Tyr motif spaced across the binding loop. The fourth cluster showed no conserved pattern and proved to contain mostly nonspecific binders.
The team synthesized 95 candidates as crude L-peptides and screened them by surface plasmon resonance. Four leads emerged with selectivity ratios favoring D-CXCL8 over L-CXCL8, confirming stereospecific binding. When synthesized as purified D-peptides, these candidates bound natural L-CXCL8 with apparent KD values ranging from 170 to 990 nanomolar. Three peptides showed clean selectivity for CXCL8 over related chemokines CXCL1, CXCL4, and CCL5. One peptide, D-2A5, bound multiple chemokines, possibly due to its three tyrosine residues mimicking receptor-binding motifs.
NMR spectroscopy revealed that the selected peptides adopt twisted β-hairpin structures stabilized by the CLIPS scaffold. When the researchers examined how these peptides affect CXCL8, they discovered something unexpected: binding caused the native CXCL8 dimer to dissociate into monomers and disrupted the chemokine's interaction with glycosaminoglycans. However, the peptides did not block CXCL8-induced cell migration in functional assays. This disconnect suggests the peptides target the GAG-binding surface rather than the receptor-binding site, offering a distinct mechanism that could complement other neutralization strategies.
The current leads require optimization before therapeutic application. Their submicromolar affinities, while respectable for initial hits, need improvement by one to two orders of magnitude. The authors point to recent success enhancing similar CLIPS peptides through incorporation of non-canonical D-amino acids, loop length variations, and alternative cyclization chemistries. This work establishes that mirror-image phage display combined with CLIPS cyclization can deliver proteolytically stable chemokine binders, opening a path toward peptide therapeutics for inflammation-driven diseases that have long resisted conventional drug discovery approaches.
Publication Information
Author Information

Dr. Ingrid Dijkgraaf studied Molecular Sciences with a specialization in Organic Chemistry at Wageningen University in the Netherlands. She then undertook joint doctoral research at the Department of Medicinal Chemistry and Chemical Biology at Utrecht University under Rob M. J. Liskamp and at the Department of Nuclear Medicine at Radboud University Medical Centre under Otto C. Boerman, developing radiolabeled RGD peptides for tumor imaging. Following her Ph.D., she was a postdoctoral fellow from 2006 to 2009 at the Department of Nuclear Medicine at the Technical University of Munich, Germany, working with Hans-Jürgen Wester and Markus Schwaiger. After a subsequent postdoctoral position at Radboud University Medical Centre, she joined the Department of Biochemistry at Maastricht University in 2011, where she is now Full Professor in Biomimetic Chemistry. In 2014, she was a Fulbright Scholar in the laboratory of Philip E. Dawson at The Scripps Research Institute in La Jolla.
Dijkgraaf's research focuses on the design and synthesis of peptides and proteins for molecular imaging, drug development, and elucidating molecular mechanisms in cardiovascular disease and oncology. Her group pursues structure-activity relationship studies of polypeptides, often drawing inspiration from hematophagous parasites—blood-feeding organisms that express an extraordinary repertoire of anticoagulant, anti-inflammatory, immunomodulatory, and vasodilating proteins to evade host defenses. These organisms represent rich sources of lead compounds for pharmacological tools and potential therapeutics. Since spring 2025, she has served as Chair of the Division of Medicinal Chemistry and Chemical Biology of the Royal Netherlands Chemical Society.

Dr. Stepan S. Denisov earned his Ph.D. in 2019 from Maastricht University, the Netherlands under supervision of Professors Ingrid Dijkgraaf and Tilman Hackeng. He then continued the work in Dijkgraaf’s group as Kootstra Talent Postdoctoral fellow. In 2022 he joined the lab of Professor Shoumo Bhattacharya at the University of Oxford as NWO Rubicon fellow. Currently Stepan is a Marie Skłodowska-Curie Postdoctoral Fellow in the lab of Professor Markus Muttenthaler at the University of Vienna. His scientific interests include (semi)synthesis and structure elucidation of cysteine-rich proteins, phage-display, protein-protein interactions and chemoinformatic.