Chiral sulfur compounds are gaining traction in drug discovery. Unlike their achiral counterparts, molecules with stereogenic sulfur centers often show improved potency and selectivity. IFM Therapeutics recently demonstrated this principle with a new NLRP3 inflammasome antagonist, the R-configured sulfonimidamide outperformed earlier achiral sulfonyl urea designs. Yet synthesizing these nitrogen-rich sulfur stereocenters remains challenging, with catalytic asymmetric methods still in their infancy.
While asymmetric oxidation of sulfur(II) compounds is well established, catalytic amination of sulfenamides has received far less attention. Previous approaches relied on stoichiometric chiral reagents or were limited to secondary amines. Scott Miller's group at Yale, published in the Journal of the American Chemical Society, set out to develop a broadly applicable catalytic solution using peptide-based organocatalysts.
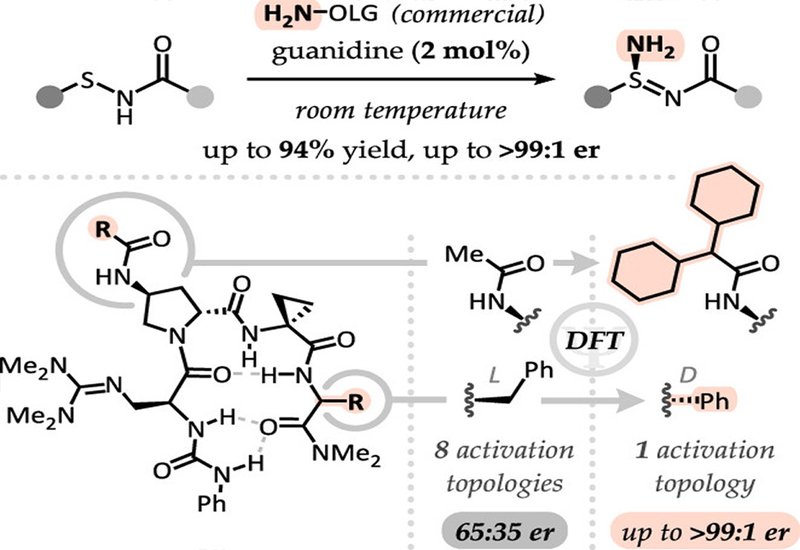
The team discovered that a commercial, bench-stable reagent (O-(4-nitrobenzoyl)hydroxylamine) cleanly transfers a single amino group to sulfenamides under mild conditions. Initial screening of chiral bases identified tetrameric peptide guanidines as promising catalysts, though selectivity was modest. Rather than relying on trial-and-error library screening, the researchers turned to density functional theory, DFT, calculations to understand why.
Computational modeling revealed that the initial catalyst allowed multiple energetically similar transition states leading to opposite enantiomers. This insight suggested a clear optimization strategy, modify the catalyst to favor a single activation mode. The team introduced bulkier substituents on one arm of the peptide, which DFT predicted would create steric clashes with undesired pathways. Experiments confirmed the prediction, with selectivity improving from 65:35 to 79:21 er.
Further computational analysis uncovered another issue, product inhibition caused by difficult catalyst turnover. The calculations guided additional modifications at the i+3 position, ultimately identifying a phenylglycine residue that both lowered the activation energy and facilitated product release. The optimized catalyst achieved selectivities up to 99:1 er across diverse substrates, including primary, secondary, and tertiary alkyl groups. A Celecoxib-derived sulfenamide converted to its sulfinamidine in 92:8 er, demonstrating the method's potential for transforming existing drugs into chiral sulfur analogs.
This work showcases how atomistic modeling can accelerate catalyst optimization for conformationally flexible peptide systems. The synergy between computation and experiment transformed a modestly selective hit into a highly effective catalyst, opening efficient access to enantioenriched sulfur(IV) building blocks for medicinal chemistry.