The misfolding and aggregation of transthyretin, TTR, a serum protein responsible for transporting thyroxine and retinol binding protein, underlies a class of amyloid diseases that include TTR cardiomyopathy and polyneuropathy. In these disorders, the native tetrameric structure of TTR becomes destabilized, dissociating into monomeric intermediates that misfold and self-assemble into soluble oligomers before progressing to insoluble amyloid aggregates. Deposits of these aggregates in the heart and nervous system lead to debilitating, often fatal outcomes. Wild-type TTR amyloidosis alone affects nearly one quarter of individuals over the age of 80, underscoring the urgent need for improved therapeutic strategies.
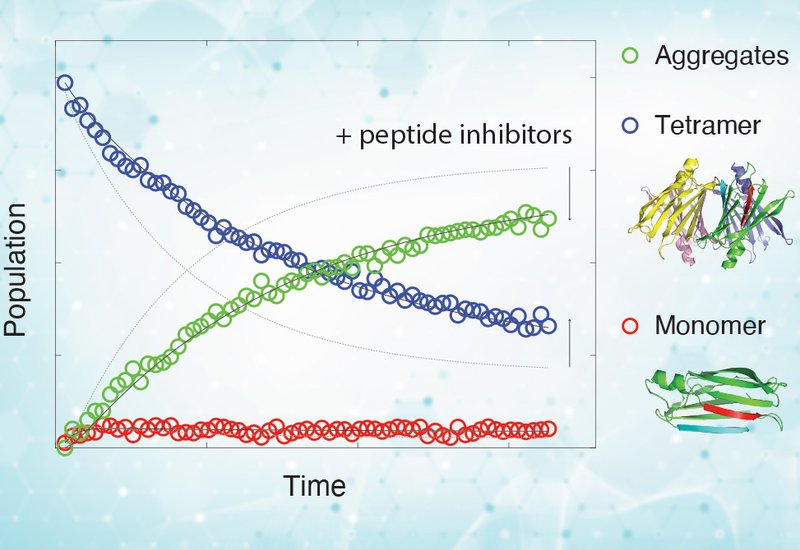
Current treatment with tafamidis, the first FDA-approved small-molecule stabilizer, works by binding to the central tetramer cavity to prevent dissociation. While effective in slowing disease progression when administered early, tafamidis is less effective in patients with advanced amyloidosis, where irreversible organ damage has already occurred. Moreover, tafamidis does not inhibit seeded aggregation, a process by which pre-existing fibrils act as templates for further amyloid growth. To address these limitations, researchers have developed short peptide inhibitors that mimic the solvent-exposed F and H β-strands of monomeric TTR. These peptides can cap aggregation-prone intermediates, yet until now, their precise effects on the individual kinetic steps of TTR aggregation remained unknown.
In a study published in the Journal of Biological Chemistry, researchers from the Peter E. Wright group at Scripps Research Institute, La Jolla, CA, in collaboration with scientists from the University of Texas Southwestern Medical Center in Dallas, TX, employed real-time 19F-NMR spectroscopy in combination with kinetic modeling to dissect how peptide inhibitors modulate the multi-step aggregation pathway of TTR. Their results provide direct mechanistic evidence that inhibitors such as TabF2, TabH2, and the concatenated construct ffTAD1 bind selectively to misfolded monomers, destabilized tetramers, and soluble oligomers at acidic pH. Importantly, these inhibitors do not interact with properly folded tetramers or monomers under neutral conditions, nor do they bind amorphous aggregates formed in vitro. Instead, they preferentially stabilize aggregation-prone intermediates, lowering the rates of tetramer dissociation and forward aggregation while reshaping the free-energy landscape of the pathway.
Interestingly, under conditions that favor the accumulation of soluble oligomers, acidic pH and reduced temperature, the peptide inhibitors also engage these oligomeric species. This observation suggests that the oligomers share structural features with patient-derived amyloid fibrils, reinforcing the therapeutic relevance of these peptide designs. By contrast, the inability of the inhibitors to bind amorphous aggregates formed in vitro highlights important conformational differences between laboratory-generated and in vivo fibrils.
Together, these findings reveal how peptide inhibitors act at the earliest stages of aggregation, providing a quantitative, step-resolved model for how they slow or halt amyloid formation. The work clarifies why these inhibitors display potent sub-stoichiometric activity: because the vulnerable intermediates are present only in low abundance, even small amounts of peptide are sufficient to exert significant inhibitory effects.
Beyond its relevance to TTR amyloidosis, the study establishes a powerful experimental and computational platform for probing aggregation kinetics in detail. By combining 19F-NMR with kinetic modeling, the approach is readily extendable to other amyloidogenic proteins—including α-synuclein, β-amyloid, and huntingtin—where similar multi-step pathways drive pathological aggregation. In doing so, it opens the door to more precise, mechanism-based design of peptide and small-molecule inhibitors targeting early misfolded states.