When Professor Maja Köhn took the stage at the 29th American Peptide Symposium in San Diego, her presentation, "Targeting Phosphatases with Peptides and Phosphomimetics," offered a captivating look into one of the most challenging frontiers in molecular medicine. Her talk built directly upon her recent publication in Biochemistry, which laid out a sweeping framework for understanding how phosphatases, those long-overlooked counterparts to kinases, are regulated, how they recognize their substrates, and how they can now be approached as druggable targets.
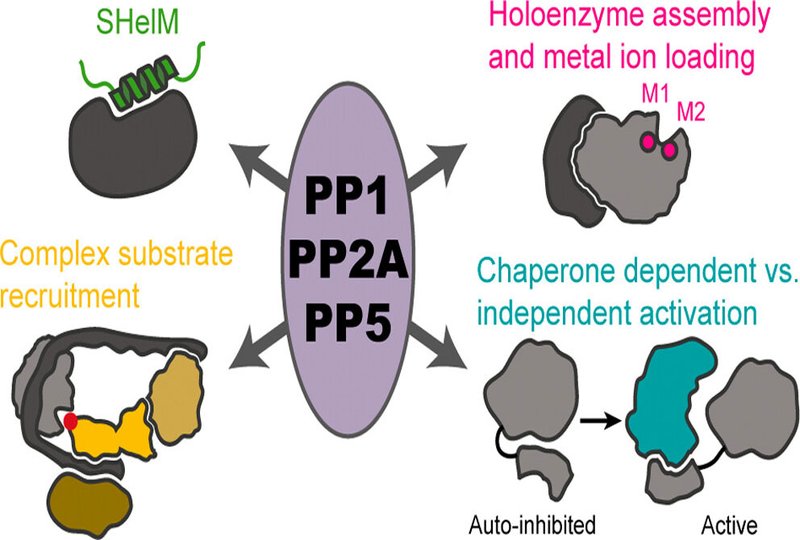
For decades, the field has wrestled with the frustrating reality that phosphatases are, by design, difficult to target. Unlike kinases, whose deep catalytic pockets lend themselves well to small molecule inhibition, phosphatases often operate through broad, shallow protein-protein interfaces. These interactions are essential for their function as holoenzymes, complex molecular assemblies in which a catalytic subunit is scaffolded by regulatory proteins. It is precisely this complexity that has made them so resistant to traditional drug discovery efforts.
Köhn’s work reframes this challenge as an opportunity, pointing to peptides, and their engineered derivatives, as uniquely suited tools for modulating phosphatase activity. Peptides are inherently capable of engaging large surface areas and can be designed to mimic, disrupt, or stabilize the kinds of transient interactions that define holoenzyme architecture. Her lab’s work has shown that it is possible not only to inhibit key phosphatase interactions but also to fine-tune them, guiding their assembly, substrate specificity, and even catalytic output.
Professor Köhn's research highlights a series of mechanistic breakthroughs that have rapidly reshaped the field. In the case of PP1, recent studies reveal that substrate recognition often extends beyond canonical short linear motifs, involving distal proteins that effectively serve as molecular scaffolds for recruitment. The discovery that the regulatory protein PPP1R15A recruits the PP1 substrate eIF2α through a two-site mechanism, one involving direct binding to the catalytic subunit, the other mediated by a distant interaction with eIF2γ, has fundamentally changed how scientists think about phosphatase targeting.
Similar paradigm shifts are unfolding in the study of PP2A holoenzymes. While it was once assumed that most phosphatase–substrate interactions were mediated by simple linear motifs, recent structural work has revealed that PP2A:B55 complexes rely on a different recognition strategy entirely, one based on short helical motifs rather than linear sequences. This discovery not only explains why earlier attempts to predict substrates from sequence data often failed but also opens the door to a new class of peptide-based inhibitors and modulators specifically designed to engage these structured interfaces.
Köhn’s group has extended these concepts beyond PP1 and PP2A to PP5, a particularly intriguing member of the phosphatase family that regulates the stress response and protein quality control through an autoinhibitory mechanism involving its tetratricopeptide, TPR, domain. Her work has contributed to the emerging understanding of how PP5’s interactions with chaperones like HSP90 and CDC37 regulate both its activation and its substrate specificity, offering yet another example of how peptide-based tools can be leveraged to dissect, and ultimately control, complex phosphatase signaling networks.
Throughout her San Diego presentation, Köhn emphasized that while phosphatases have traditionally been viewed as intractable targets, the problem has always been one of tool design, not biology. Peptides, with their inherent ability to bridge large, complex protein surfaces, are not simply workarounds for difficult targets—they are, in many cases, the ideal modality for interrogating and manipulating the dynamic assemblies that define phosphatase function.
Her lab’s work in designing phosphomimetics, photoactivatable peptides, and PPI disruptors underscores this point. These tools have revealed unexpected insights, including cases where binding affinities or biochemical outcomes diverge from classical predictions. Such surprises are not setbacks but rather opportunities, offering deeper understanding of the fluid, adaptable logic by which these enzymes operate within the cell.
It was particularly fitting that this talk came on the heels of her landmark 2025 publication, as the two formed a seamless narrative arc. The paper laid the conceptual and structural groundwork; the San Diego presentation brought it vividly to life. Together, they reflect a field not merely advancing incrementally but undergoing a profound conceptual transformation. What was once deemed undruggable is now revealed to be exquisitely tunable—provided the right molecular language is spoken.
In Köhn’s hands, that language is peptide-based, structurally precise, and chemically sophisticated. Her work stands as a compelling demonstration that the future of phosphatase biology—and perhaps of many other so-called "undruggable" targets—will be written not in small molecules alone, but in the versatile and expanding vocabulary of peptides and phosphomimetics.