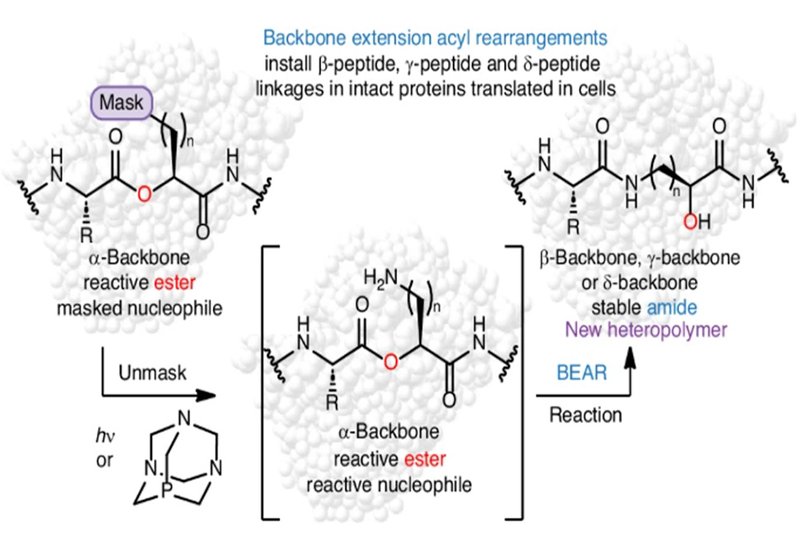
The promise of proteins whose backbones deviate from the canonical α-architecture has been clear for decades: a few strategically placed methylene units can toughen a chain against proteolysis, reshape cell permeability, retune major histocompatibility complex (MHC) presentation, or reorganize a binding surface to modulate protein–protein interactions. Yet despite that promise, reliably installing extended-backbone linkages—β, γ, or δ—in cells has remained frustratingly rare. The obstacles are familiar—engineered monomers strain EF-Tu delivery, challenge the ribosome’s peptidyl-transferase chemistry, and often depend on stoichiometric tRNA acylation tactics that do not port readily to living systems. In a study published in Nature Chemical Biology by researchers in the Alanna Schepartz and Matthew B. Francis groups at the University of California, Berkeley, in collaboration with colleagues from the Scott J. Miller group at Yale, the authors sidestep those constraints by reframing backbone engineering as a post-translational problem. Rather than forcing the ribosome to stitch in a β/γ/δ unit, they embed a latent rearrangement into a ribosomal product and let the protein edit itself. Their work was accompanied by a Nature Chemical Biology Research Briefing that highlights the broader significance of this strategy.
The centerpiece is the Backbone Extension Acyl Rearrangement, BEAR, an intramolecular acyl shift analogous to the second step of native chemical ligation. During translation in E. coli, an orthogonal pyrrolysyl-tRNA synthetase (PylRS)/tRNA pair installs α-hydroxy-acid analogs whose side chains carry masked nucleophiles. After expression, the mask is removed—photochemically or chemically—to reveal a tethered amine that is preorganized to attack the neighboring O-acyl linkage. The result is a spontaneous O→N acyl transfer that both forges an amide and adds methylene unit(s) to the main chain, yielding a site-specific β2, γ, or δ backbone within a full-length, genetically encoded protein.
Density functional theory provides the first indication that such edits should work. Across model systems corresponding to the β, γ, and δ cases, BEAR rearrangements are thermodynamically favored relative to canonical cysteine/serine NCL analogs by roughly 2–3 kcal·mol−1, an advantage traced to stabilizing hydrogen bonds in the products and relief of steric congestion. The intermediates pay a higher entropic price—especially for the seven-membered ring en route to a δ linkage—foreshadowing slower kinetics for δ relative to β or γ. Those expectations map cleanly onto the experimental data.
To test feasibility in a protein context, the team first targets a single site in NanoLuc with a photomasked α-hydroxy isoserine (pm-isoS) that can unveil a β-amine upon irradiation and thereby trigger a β2-forming O→N shift. Installation via a PylRS variant proceeds in cells; controlled photolysis then drives the rearrangement. Intact-protein LC–MS confirms correct mass, but the decisive evidence comes from peptide-level co-elution with a synthetic standard containing the anticipated β2 linkage and indistinguishable MS/MS fragmentation—a rigorous fingerprint that the backbone truly has been extended at the programmed site. A practical note emerges here: executing the unmasking after purification improves conversion and reduces hydrolysis and background, yielding cleaner maps than in-cell light exposure.
Having established the principle, the authors pivot to a γ-extension, using γ-N3-OH, an azide-bearing α-hydroxy acid that can be reduced to a γ-amine and then allowed to rearrange. Expression in SUMO–GFP is robust, notably in C321.ΔA.exp cells, and the purified protein again matches the expected mass. The reduction chemistry matters: commonplace phosphines such as TCEP or THPP generate significant alcohol side product from azide, diminishing the desired rearrangement. In contrast, 1,3,5-triaza-7-phosphaadamantane (PTA) cleanly unmasks the amine with minimal diversion, and under mildly basic conditions, pH 8.2; 0 °C then 42 °C, the intramolecular O→N transfer proceeds to near-quantitative yield with >90% fidelity at the edited position. One mechanistic nuance is instructive: at elevated pH in the absence of a reductant, ester hydrolysis competes, inflating apparent misincorporation in mapping; driving BEAR promptly after unmasking solves that problem by replacing the labile ester with a stable amide.
The most challenging case is the δ-extension, which must traverse a higher-energy, seven-membered intermediate. Here, the group incorporates δ-N3-OH at a defined site in SUMO–GFP, purifies the single-site variant, and explores conditions that favor the rearrangement after azide reduction. As predicted by theory, time and temperature are the levers: extended incubations, PTA, pH 8.2, 50 °C, 48 h, convert the reduced intermediate into the δ-linked product in ~27% yield, with hydrolysis as the principal competitor under the harsher conditions. The outcome is still a landmark: a site-specific δ linkage formed inside a folded, full-length protein expressed in cells, achieved without ribosome or EF-Tu engineering.
Methodologically, BEAR is notable for what it doesn’t require. There is no need to retool the ribosome’s active site or tune EF-Tu to carry non-standard cargo. The only special machinery is an orthogonal aaRS/tRNA pair capable of accepting α-hydroxy acids that bear maskable nucleophiles. From there, chemistry takes over: unmask the side chain, allow the preorganized intramolecular attack, and capture the extended backbone as a standard amide. The strategy is modular, choice of mask, trigger, and nucleophile, temporal, unmasking can be staged in cells or after purification; photochemistry offers spatial control, and general, the same logic should translate to other nucleophiles and caging groups, broadening the palette beyond the β/γ/δ exemplars.
The implications for peptide and protein science are immediate. A few judiciously spaced β/γ/δ edits can harden a fold against proteases, reshape dynamics at a ligand interface, or dial in cooperative assembly to yield protein-like materials with enhanced thermal stability. For chemical biology, BEAR offers precision backbone editing for mechanistic probes—altering H-bond geometry, backbone length, or conformational bias at a single site without changing side-chain identity. In translational contexts, one can imagine PPI modulators that exploit γ- or δ-spacing to access otherwise unreachable pharmacophores, or antigens whose MHC presentation is tuned by a backbone extension to rebalance immunogenicity. And because the edits are encoded at the gene level and executed inside cells, scale and evolvability—two chronic weaknesses of purely synthetic routes—come back into play.
There are, of course, boundaries to push. The δ case underscores that kinetics and local structure matter; sequence context, folding constraints, and microenvironment will influence rearrangement rates and competing hydrolysis. Expanding the aaRS specificity space for alternative masked monomers will widen the design window, even if the ribosome and EF-Tu remain blissfully unmodified. And the most exciting frontier is likely spatiotemporal control in living cells: photomasks and bioorthogonal triggers that confine backbone edits to defined subcellular locales or time windows, turning BEAR into a tool for dynamic proteome engineering.
In sum, BEAR converts a long-standing encoding challenge into a tractable chemical edit. By programming a post-translational acyl rearrangement into a ribosomally produced chain, the authors show that β2, γ, and δ linkages can be installed site-selectively in full-length proteins expressed in vivo, with rigorous peptide-level validation. It is a conceptually clean and practically flexible framework—one that should accelerate the creation and evolution of extended-backbone protein hybrids for basic science, materials design, and next-generation therapeutics.