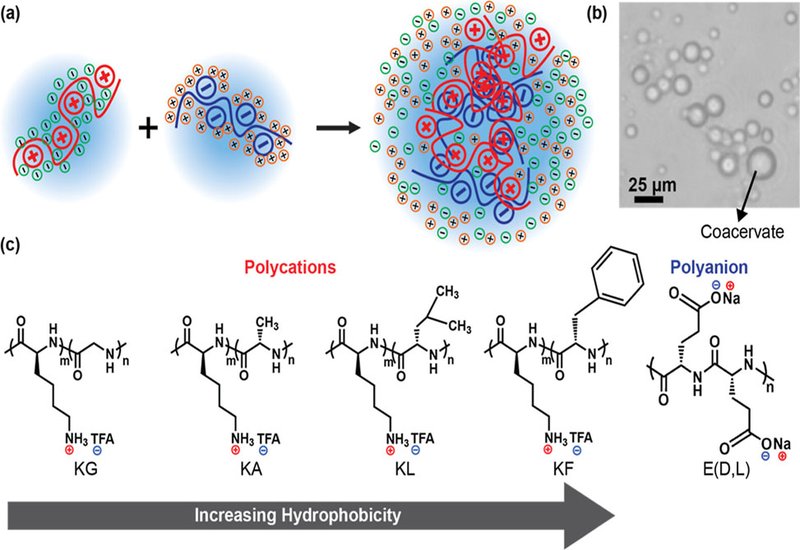
Cells routinely organize biochemical reactions within membraneless organelles formed by liquid–liquid phase separation, LLPS, where intrinsically disordered proteins, IDPs, condense into dynamic, protein- and RNA-rich droplets. Dissecting how specific sequence features encode this behavior remains a central challenge. In this study, published in Biomacromolecules, researchers in Sarah Perry's group at the University of Massachusetts - Amherst, employ sequence-defined polypeptides as minimal models of IDP-like LLPS, using complex coacervation between oppositely charged peptides to map how charge patterning and hydrophobic residues jointly tune both phase stability and viscoelastic properties.
The authors synthesized families of lysine-containing cationic peptides in which the size of contiguous charged blocks, denoted by τ, was precisely controlled, and paired these with a homopolyglutamate polyanion. Coacervation was maximal near charge neutrality, and the stability of the condensed phase was quantified by salt resistance at low polymer concentrations. Increasing τ consistently elevated salt resistance, consistent with counterion-release entropy as a principal driver of complexation. Larger runs of charge confine counterions more tightly in the dispersed state; their cooperative release upon complex formation yields a greater entropic gain, stabilizing the dense phase. These results echo sequence-encoded electrostatic patterning effects that have been inferred for cellular IDPs.
To separate electrostatics from hydrophobic contributions, otherwise identical peptide sets were prepared in which neutral glycine was replaced by alanine. Across matched τ values, alanine-containing sequences produced coacervates that withstood higher salt, indicating an additional stabilizing contribution commonly attributed to the reorganization of water around hydrophobic side chains. In this view, the condensed phase benefits not only from counterion liberation but also from the partial release and restructuring of ordered water, broadening the two-phase region even when overall charge content is unchanged.
How these sequence encodings translate into material behavior was probed by particle-tracking microrheology. For glycine-based series, viscosity rose with increasing τ, as expected for networks strengthened by longer ionic domains and more cooperative electrostatic cross-linking. In contrast, alanine-based series displayed an inverted trend in which shorter neutral blocks yielded higher viscosities than longer ones. Circular dichroism measurements resolved this apparent paradox: short-block alanine peptides retained pronounced polyproline II–like character within the coacervate, whereas longer blocks shifted toward mixtures suggestive of random coil and α-helix. The emergence of PPII structure correlates with increased chain stiffness and higher local charge density, both of which can raise viscosity despite identical nominal composition. Thus, secondary structure provides an additional, sequence-sensitive axis by which peptide condensates modulate mechanics, complementing electrostatics and hydrophobicity.
The design space was then extended by varying neutral residue hydrophobicity (G < A < L < F) while maintaining overall chain length and introducing higher-charge motifs (K6X2 and K3X). With increasing hydrophobicity, coacervates exhibited systematically greater salt resistance and dramatic viscosity gains, culminating in phenylalanine-rich systems that approached elastic-dominant behavior. The storage and loss moduli crossed over as hydrophobic content increased, indicating a transition from viscous liquid to viscoelastic gel–like response. Concomitantly, the power-law exponent from microrheology decreased with hydrophobicity, consistent with heightened heterogeneity and clustering within the dense phase. These observations align with intuitive expectations for hydrophobe-driven association, yet are now demonstrated in a sequence-controlled peptide platform rather than in random copolymers.
Taken together, the work establishes clear molecular links between sequence patterning, phase stability, and rheology in peptide-based coacervates. Charge-block architecture dictates the magnitude of counterion-release entropy and thereby the robustness of phase separation to salt; hydrophobic residues add an orthogonal stabilization pathway through water-structuring effects and promote elasticity by densifying associating domains; and specific patterns can bias secondary structure formation inside the condensed phase, shifting chain stiffness and network connectivity in ways not predicted by composition alone. The net effect is a tunable palette of condensate behaviors—from fluid droplets to stiffer, more elastic materials—encoded directly in amino acid sequence.
For biological condensates, these findings support a mechanistic picture in which IDP motifs leverage coupled electrostatic and hydrophobic codes, together with sequence-dependent backbone preferences, to control the balance between liquidity and solidity as function demands. For materials design, the study provides actionable rules for engineering peptide coacervates with targeted salt tolerance and mechanical response by adjusting charge-block size, neutral-residue identity, and their patterning along the chain. By marrying microrheology with controlled sequence chemistry, Perry’s team delivers a compact framework for translating molecular edits into emergent condensate function—advancing both the understanding of IDP physics and the development of programmable, biomimetic soft materials.