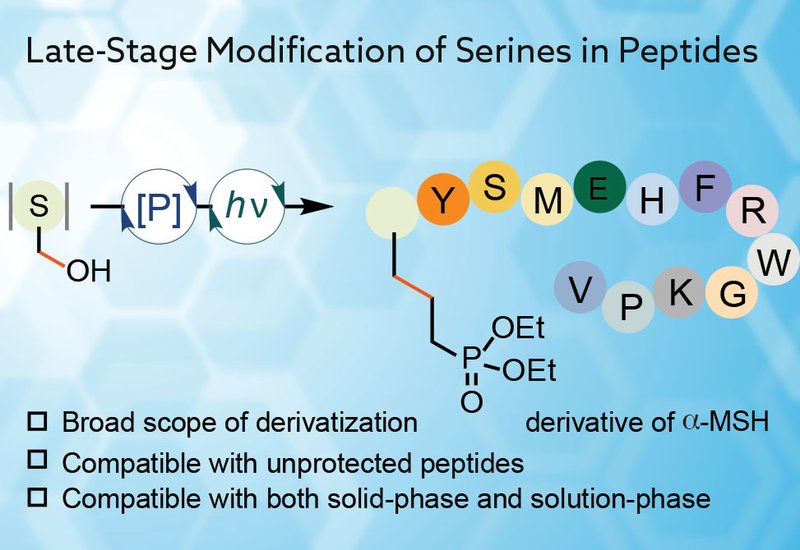
Published in the Journal of the American Chemical Society, work by first author Zhenyan Guo in the Diao Lab at the New York University, delivers a chemoselective solution to a long-standing gap: late-stage modification of native serine via carbon–carbon bond formation in complex peptides. The authors adapt phosphoramidite chemistry, famous for automated DNA synthesis, to activate the serine hydroxyl under mildly acidic conditions, then transform it through a photoredox deoxygenative coupling to diverse alkene partners. The method represents a general platform that converts a native Ser residue into a broad panel of noncanonical side chains, such as homoglutamine, homoglutamate, 5-hydroxynorvaline, and phosphonates, and even isotopologues, Ala-3-d1, without preinstalling handles or protecting the rest of the peptide.
Mechanistically, a tethered-iodoaryl phosphoramidite 1 cleanly phosphitylates Ser to phosphite 3 with 5-methyl-tetrazole. Under 390 nm light irradiation, a strongly reducing photocatalyst, E* ≈ −2.5 V vs SCE, reduces 3 to an aryl radical that cyclizes into phosphorus; β-scission then cleaves the Ser C–O bond, generating an α-alanyl carbon radical. That radical engages Giese additions to many acceptors and is terminated by hydrogen atom transfer from IDM-BH3, validated by exclusive D-incorporation when IDM-BD3 is used. Oxygen suppresses the reaction; light and photocatalyst are required; Stern–Volmer data identify the Ser-phosphite as the primary quencher. Importantly, chiral HPLC shows no α-epimerization—this is stereoretentive at α-C. A small competing path to dehydroalanine is observed but manageable.
Scope is conspicuously broad for a late-stage peptide method. Electron-poor alkenes, vinyl nitriles, acrylamides, acrylates, vinyl phosphates, acrylic acid, give homologated side chains such as homoglutamine and homoglutamic acid; vinyl pyridine and α,α-disubstituted alkenes are tolerated; vinyl boranes can be funneled to 5-hydroxynorvaline after oxidation. Even strained or less activated partners, [1.1.1]propellane and phenylacetylene via downstream arylation, participate. The platform also enables direct installation of affinity/fluorophore handles, either by using prefunctionalized acrylates, biotin, or by clicking downstream on an introduced handle, for example, turning an acrylate adduct into a fluorescent probe.
Selectivity is a highlight. In head-to-head competition, Ser is phosphitylated approximately four times faster than Thr; Tyr and Lys do accept phosphorylation but their intermediates do not undergo the deoxygenative coupling and simply hydrolyze back, restoring the native residues; Cys is not phosphitylated and does not interfere with the photo reaction. That orthogonality lets the chemistry run cleanly in unprotected, polar peptide settings.
The method works both on solid support and in solution. On-resin, a TBS-protected Ser is unveiled and immediately taken through activation and coupling to give, for example, a diethyl phosphate–modified Ser in an overall 32% isolated yield from resin across a standard SPPS sequence. In solution, the same transformation proceeds in comparable overall efficiency, for example, 38% from the isolated peptide, with partial conversion at the phosphorylation. Swapping the alkene for IDM-BD3 with no acceptor provides Ser→Ala-3-d1, useful for pharmacokinetic probes or radiotracer analog development.
Compatibility is evidenced across pharmacologically and biologically relevant peptides: enkephalin, bradykinin, and α-MSH are all modified cleanly at Ser. An atosiban analog is accessed by intramolecular capture, N-terminal acrylate, to induce macrocyclization, illustrating on-peptide conformational editing. Across these examples, hydrophilic (Arg, His, Lys, Glu, Thr, Asn) and hydrophobic (Gly, Val, Ile, Met, Phe, Pro, Tyr, Trp, Cys) side chains are tolerated without bespoke protection.
The significance is twofold. First, it finally enables late-stage Ser editing by C–C bond formation under conditions that respect native peptide complexity—ideal for SAR libraries and medicinal chemistry across lead peptides, macrocycles, and probes. Second, it introduces an orthogonal, modular entry to noncanonical residue space from a native amino acid, with stereochemical fidelity and programmable handles. Practical considerations remain, photoredox setup; exclusion of O2; managing minor Dha formation; phosphorylation efficiency in highly polar sequences, but the operational simplicity, chemoselectivity, and breadth make this a workhorse method for peptide diversification and tool compound assembly.