Selective modification of amino acid side chains has become a central strategy for expanding peptide function beyond the limits of ribosomal chemistry. Yet one residue remains notably resistant to this creativity. Arginine, which contributes nearly six percent of residues in proteins, carries a guanidinium group with high pKa and low nucleophilicity, properties that make it indispensable for molecular recognition but difficult to transform cleanly. Existing arginine-labeling reagents often generate complex mixtures, limiting their use in peptide engineering. Developing a robust approach that can target arginine directly, without collateral reactivity, remains a major challenge for chemical biology.
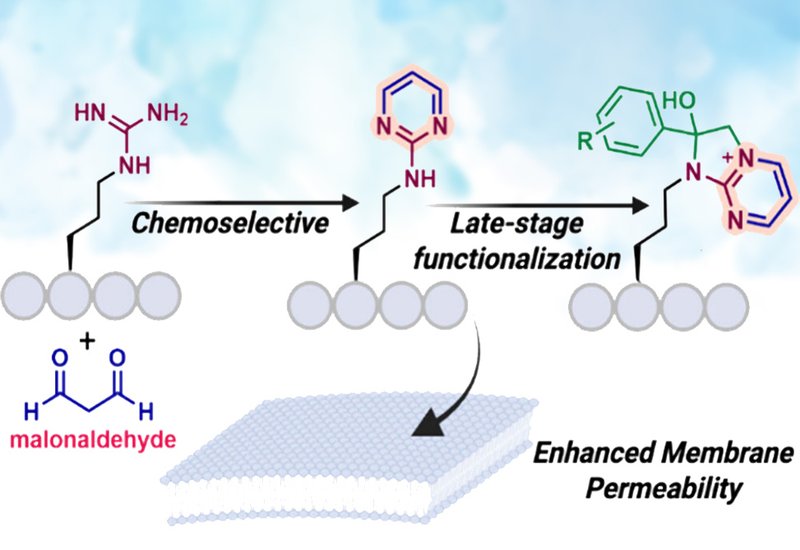
To address this gap, researchers in the Raj group at Emory University, published in Organic Letters, designed an acid-mediated strategy that converts arginine’s guanidinium into a stable amino pyrimidine ring. The method uses malonaldehyde to achieve selective cyclization under strong acidic conditions, producing the heteroaromatic product with near-quantitative efficiency across diverse peptide substrates. Crucially, condensation side products from nucleophilic residues such as lysine, tryptophan, or histidine can be reversed with butylamine, restoring a single arginine-derived product. This combination of chemoselectivity, reversibility, and high conversion marks a substantial advance in peptide modification.
Reaction optimization revealed that malonaldehyde reacts cleanly with arginine only under highly acidic conditions. While aqueous, basic, and buffered media produced no detectable products, 12 M HCl enabled rapid and efficient conversion, reaching more than ninety-nine percent with excess malonaldehyde. The protocol tolerated a wide range of peptide sequences, including substrates containing multiple arginine residues, which were converted quantitatively to their amino pyrimidine forms. Peptides bearing other reactive residues generated predictable condensation byproducts, all removable with butylamine. Notably, in peptides lacking arginine, histidine showed only trace reactivity and cysteine remained unmodified, underscoring the method’s functional selectivity.
The transformation also has consequences for biological function. Cell-based permeability assays demonstrated that converting arginine to the amino pyrimidine form increased membrane uptake by approximately twofold. This enhancement likely arises from redistribution of positive charge within the heterocycle and a modest gain in hydrophobicity, properties that facilitate more efficient passage across cellular membranes. These observations suggest that the modification acts not only as a chemical handle but as a functional enhancer with direct implications for peptide delivery.
Beyond simple labeling, the amino pyrimidine products support late-stage diversification. Reaction with substituted 2-bromoacetophenones generated fused imidazo[1,2-a]pyrimidinium salts in good yields, expanding the structural space accessible from a single arginine residue. These heteroaromatic motifs are widely used in medicinal chemistry, offering opportunities for tuning polarity, stability, and binding interactions. Because the transformation proceeds under mild conditions and preserves peptide integrity, it is well suited for modular construction of peptide-based drug candidates.
Together, this chemistry reframes arginine not as a difficult residue but as an entry point into heteroaromatic diversification. By enabling efficient formation of amino pyrimidines, reversible cleanup of side reactions, improved cellular permeability, and access to fused ring systems, the method broadens the functional possibilities of peptides. It offers a practical route to enhance bioavailability and pharmacological behavior while preserving the sequence-encoded specificity that makes peptides valuable therapeutic scaffolds.