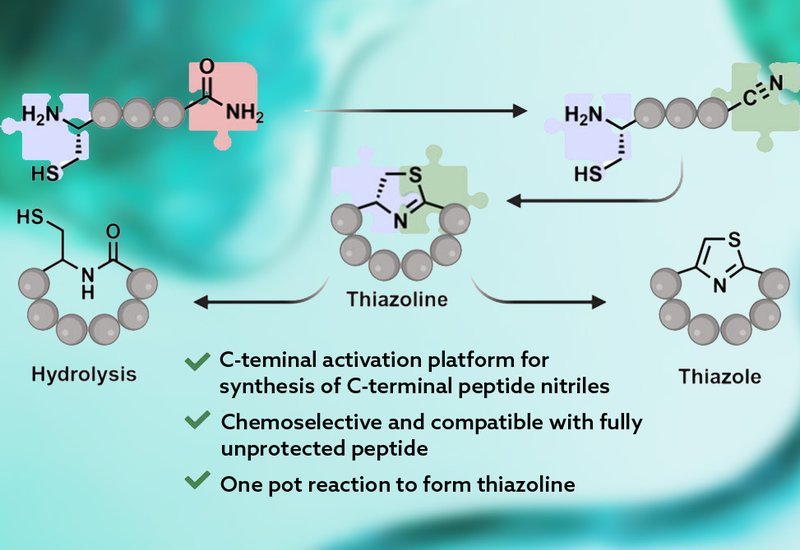
Macrocyclic peptides occupy a valuable middle ground between small molecules and biologics, offering conformational constraint, membrane permeability, and proteolytic stability that neither class easily achieves alone. Marine-derived natural products such as Mollamide F, Sanguinamide A, and Haligramide A exemplify one particularly potent architectural strategy: thiazoline and thiazole heterocycles embedded directly within the peptide backbone. Unlike side-chain staples or external cross-links, these backbone-embedded rings restrict conformational motion and mask polar amide bonds, but accessing them synthetically has remained a persistent challenge. Classical cyclodehydration routes can be harsh and substrate-dependent, while more recent N-terminal cysteine condensation methods require presynthesized chiral α-amino nitrile building blocks, a step that lengthens library preparation and introduces epimerization risk, particularly for residues bearing reactive side chains.
Researchers in the Raj Group at Emory University, published in Organic Letters, developed a late-stage, sequence-tolerant C-terminal activation strategy that bypasses the need for specialty nitrile building blocks entirely. The platform uses palladium(II) trifluoroacetate, Pd(TFA)2, to selectively dehydrate the C-terminal primary amide of a fully assembled, unprotected linear peptide to the corresponding nitrile at room temperature in acetonitrile. Because the transformation acts on a completed peptide rather than on individual amino acid derivatives, stereochemical integrity at all chiral centers is preserved. Dithiothreitol, DTT, then scavenges residual palladium as an insoluble Pd-thiol complex, freeing the C-terminal nitrile for subsequent N-terminal cysteine condensation and cyclization in a 1:1 isopropanol/phosphate-buffered saline mixture at pH 7.4.
Chemoselectivity studies using the nine-residue peptide YWRMCKEHS showed no detectable side-chain modifications by LC-MS under the dehydration conditions, consistent with preferential reactivity toward the terminal primary amide. A key selectivity question arose because Pd(TFA)2 can also convert asparagine and glutamine side-chain amides to nitriles. To probe whether aliphatic side-chain nitriles might compete in the cyclization step, the team prepared two peptides: CWPAYA bearing a C-terminal primary amide and CWPAYQ bearing a glutamine residue and a C-terminal carboxylic acid. After Pd(TFA)2 treatment, only the peptide carrying the C-terminal backbone nitrile cyclized to the corresponding thiazoline macrocycle; the glutamine-derived side-chain nitrile instead formed a cysteine disulfide dimer with no detectable thiazoline. Notably, the asparagine/glutamine nitrile modification proved reversible: addition of Pd(TFA)2 with acetamide in a THF/water or isopropanol/water mixture restored the native amide side chain.
Applying the optimized sequence to six peptides spanning 5- to 9-amino-acid ring sizes and diverse C-terminal residues, the team achieved 95–99% conversion to thiazoline macrocycles, with the sterically hindered isoleucine derivative requiring 70 °C rather than room temperature. Subsequent oxidation with MnO2/K2CO3 delivered thiazole macrocycles in quantitative conversion for most substrates, including Sanguinamide A and Haligramide A. For substrates bearing His or Trp residues, MnO2-free conditions using K2CO3 alone furnished the thiazole products in high conversion without side-chain oxidation. A final formic acid hydrolysis step converted thiazoline macrocycles back to native-backbone macrocycles bearing a cysteine residue at the former cyclization site, demonstrating bidirectional access to three distinct macrocyclic topologies from a single linear precursor.
The platform collapses what was previously a multistep building-block synthesis problem into a one-pot late-stage activation on fully deprotected peptides, and the total syntheses of Mollamide F, Sanguinamide A, and Haligramide A from their linear amide precursors illustrate the practical reach of the method. Compatibility with reactive side chains including Cys, Arg, Trp, His, Ser, Tyr, Lys, Glu, and Met positions the approach as a general tool for generating thiazoline/thiazole macrocycle libraries directly from SPPS-derived linear peptides. The reversibility of side-chain nitrile formation and the ability to intercept the thiazoline intermediate for either oxidation or hydrolysis offer additional synthetic flexibility for exploring backbone-heterocycle diversity in peptide drug discovery.