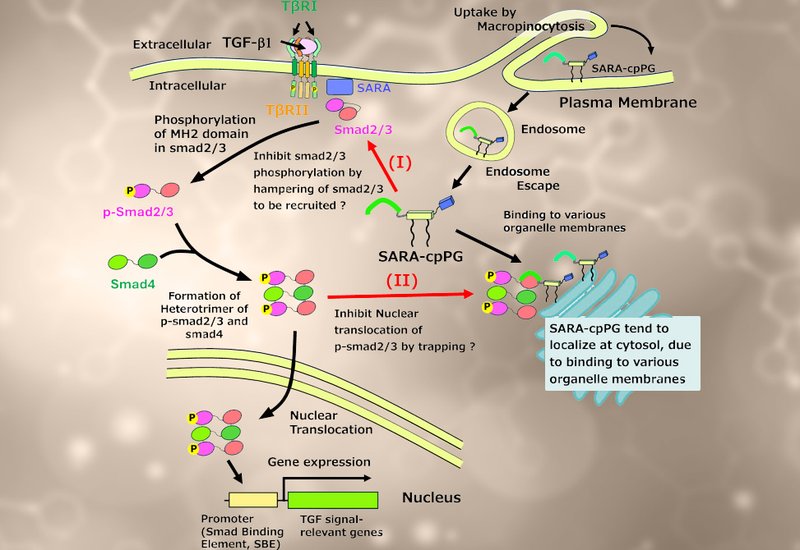
Peptide-based therapeutics hold enormous promise for modulating intracellular signaling, but their impact has been limited by poor membrane permeability. Most peptide drugs remain confined to extracellular or cell-surface targets, leaving a vast landscape of cytosolic and nuclear signaling proteins largely out of reach. Among the most consequential of these intracellular targets is Smad2/3, a central mediator of transforming growth factor-beta, TGF-β, signaling that travels from the cytoplasm to the nucleus to drive transcription of genes promoting cell migration, epithelial-mesenchymal transition, EMT, and cancer progression. Blocking this nuclear journey, rather than suppressing upstream kinase activity, offers a mechanistically distinct and potentially more selective therapeutic strategy.
Researchers in the Mizuno Group at Nagoya Institute of Technology, published in Bioconjug. Chem., developed a conjugate strategy to deliver a Smad2/3-binding peptide directly into the cytoplasm using a cell-penetrating peptide-gemini surfactant, cpPG, carrier designated DKDKC12-K5. The cargo peptide derives from SARA, Smad anchor for receptor activation, a protein whose Smad-binding domain makes direct contact with the MH2 domain of Smad2. To enable conjugation, the team replaced an internal cysteine in the SARA sequence with serine and appended a new cysteine via a flexible linker at the C-terminus, then coupled this modified SARA peptide to Mal-DKDKC12-K5 through a maleimide-thiol Michael addition reaction. The resulting conjugate, SARA-cpPG, was tested in A549 human lung adenocarcinoma cells, a well-established model for TGF-β-driven EMT.
Cellular uptake of SARA-cpPG exceeded that of the SARA peptide alone by more than 20-fold, and confocal microscopy confirmed that the conjugate localized predominantly in the cytoplasm, with no detectable redistribution to the nucleus. This cytoplasmic retention proved functionally critical. TGF-β1 stimulation normally drives a sharp increase in A549 cell migration and pronounced actin fiber polymerization; SARA-cpPG suppressed both responses to levels at or below untreated controls, performance comparable to LY2157299, a small-molecule kinase inhibitor of the TGF-β type I receptor. A control conjugate carrying a mutant SARA peptide with abolished Smad2/3 binding affinity, SARAm-cpPG, showed negligible inhibition, confirming that the effect depends on direct Smad2/3 engagement. Mechanistic studies clarified the point of intervention: Western blot analysis showed that SARA-cpPG left Smad2 phosphorylation fully intact, ruling out upstream kinase interference. A luciferase reporter assay driven by the Smad-binding element confirmed that SARA-cpPG suppressed TGF-β-responsive gene expression, consistent with a blockade of phosphorylated Smad2/3 nuclear translocation rather than inhibition of its activation.
The work establishes DKDKC12-K5 as a delivery platform with a decisive advantage over conventional carriers such as the R8 octa-arginine peptide, which entered cells but failed to suppress TGF-β signaling, likely because it lacked cytoplasmic retention and permitted co-transport of the SARA peptide into the nucleus alongside its target. The findings open a broader design principle: for peptide inhibitors targeting transcription factors or other signaling proteins that must be sequestered in the cytoplasm to be neutralized, cytoplasmic localization of the carrier is not incidental but essential. Validation in primary cells and additional cancer cell lines is planned, and the approach is expected to generalize to other nuclear-translocating signaling molecules.