Classical synthetic macrocycles such as cyclodextrins, cucurbiturils, calixarenes, and pillar[n]arenes have driven supramolecular chemistry for decades, yet each platform carries persistent liabilities: limited water solubility at pharmaceutically relevant cavity sizes, restricted access to inner-cavity functionalization, and synthetic routes that rely on condensation chemistry or enzymatic transformations that constrain structural diversity. Achieving the kind of positional control over both exo- and endo-functional groups that enzymes exercise over their active sites has remained an open problem in the field.
Researchers in the Palma Group at University College Dublin and the Fantuzzi Group at the University of Kent, published in Angewandte Chemie International Edition, report a solution built entirely from peptide chemistry. The team used Fmoc-based SPPS to assemble a linear hexadecameric precursor comprising alternating tetramers of L- and D-proline, then closed the chain via head-to-tail macrocyclization with PyBOP in dilute acetonitrile at 60 °C. After reverse-phase preparative HPLC, the target cyclo-polyproline, CP[4,4], where the indices denote the length of each homochiral segment, was isolated in 43% yield over the two steps.
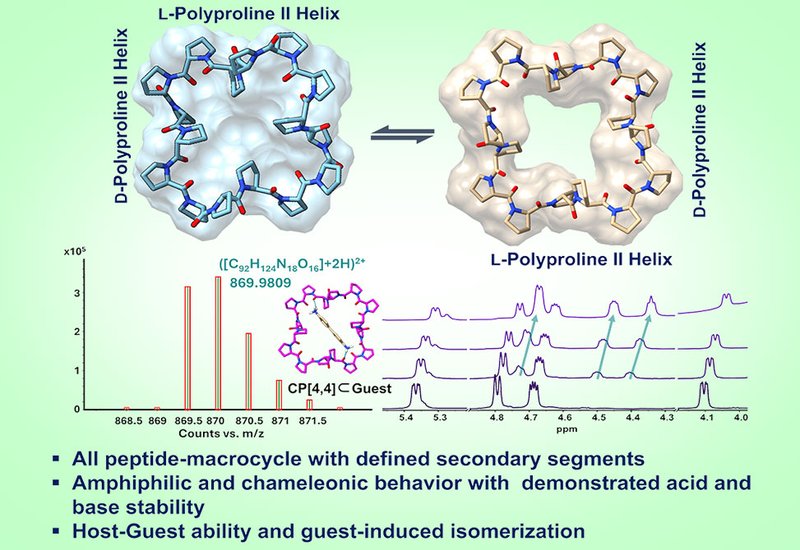
Solution NMR and single-crystal X-ray diffraction, SC-XRD, established that CP[4,4] behaves as a conformational chameleon. In CDCl3 and CD3OD, the macrocycle adopts exclusively the all-junctions-cis amide conformation at the four L/D junction points, while the internal polyproline II helical structure of each tetrameric segment is preserved. In D2O, an equilibrium mixture of roughly 60% all-junctions-trans and 40% all-junctions-cis isomers is observed, and SC-XRD crystals grown from water confirm the expanded, saddle-shaped all-junctions-trans structure. Variable-temperature 1H NMR up to 90 °C showed no peak coalescence, placing the isomerization barrier above 18 kcal mol−1 and confirming the thermal stability of the scaffold. The macrocycle proved soluble across this full range of solvents, a property that distinguishes it from many native macrocyclic hosts.
Quantum-chemical calculations at the PBE0-D3(BJ)/def2-TZVPP level, with implicit CPCM/SMD solvation, showed that the all-junctions-cis isomer is intrinsically more stable by 8.0 kcal mol−1 in chloroform, yet the balance tips in water through a cooperative hydration effect. SC-XRD of the aqueous crystals revealed four water molecules occupying well-defined pockets at the corners of the all-junctions-trans macrocycle, each forming two-point hydrogen bonds to alternating amide carbonyls with individual stabilization energies of approximately 23.2 kcal mol−1 per water molecule. Incremental addition of the second, third, and fourth water molecules delivers cumulative stabilization reaching 77.3 kcal mol−1 for the tetrahydrate complex. A 100 ns molecular dynamics simulation confirmed that the all-junctions-trans conformation persists throughout, with a recurring water bridge between intercalated proline residues separated by an oxygen–oxygen distance of approximately 4 Å.
CP[4,4] in the all-junctions-trans form presents an accessible cavity of 97.6 Å3, with a hydrophobic cleft flanked by two hydrogen-bond acceptor corners. Molecular docking with AutoDock Vina identified protonated benzidine salt 2Cl as a complementary guest, and 1H NMR titration in D2O confirmed host–guest complexation in a fast-exchange regime with an apparent association constant of 70.82 ± 3.99 M−1. The more telling result came from titrating the organic-soluble salt 2TFA into a CDCl3 solution containing exclusively the all-junctions-cis isomer: new resonances diagnostic of the all-junctions-trans form appeared, confirming guest-induced conformational switching analogous to an enzyme induced-fit mechanism. No complexation was detected with unprotonated guest 2. Mixing CP[4,4] with Fmoc-capped polyproline tetramer 3 in a 1:1 ratio in D2O produced diagnostic chemical-shift perturbations in both host and guest signals, and LC-HRMS confirmed formation of the desired complex, constituting an all-peptide pseudo-rotaxane.
CP[4,4] establishes SPPS as a viable and modular route to synthetic macrocyclic hosts, providing atom-level sequence control and a clear path to both exo- and endo-functionalization that classical platforms cannot match. The amphiphilicity, pH stability across the range 0.8 to 11.6, and solvent-switchable conformation of this scaffold position it as a starting point for programmable supramolecular hosts, potential organocatalysts, and enzyme mimetics. The authors note that the methodology is directly extendable to mixed-sequence proline-rich macrocycles carrying diverse functional groups, with applications in separation science and catalysis to be reported in future work.