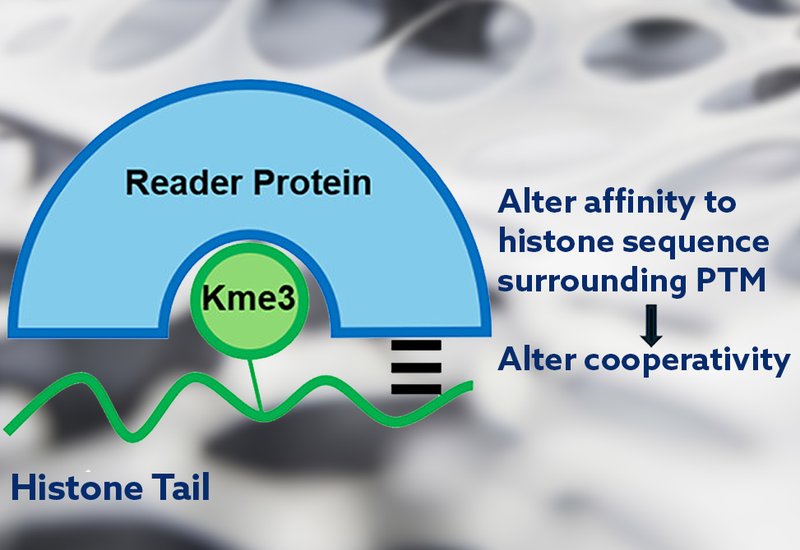
Histone trimethyllysine, Kme3, reader proteins regulate gene expression by binding to histone tails containing both the Kme3 post-translational modification, PTM, as well as the correct surrounding sequence, suggesting that the interaction is highly cooperative. This class of reader proteins includes numerous validated cancer targets, yet no approved therapeutics exist for this entire protein class. A better understanding of the role of cooperativity in the binding mechanism has the potential to advance the field.
Christopher Travis, and colleagues in Marcey Waters' laboratory at the University of North Carolina at Chapel Hill, published in Biochemistry, devised an elegant strategy to probe the cooperativity of binding through mutation of both the reader protein or histone in three model proteins. The team exploited an unusual finding: approximately 5% of human Kme3 readers bind equally well or better to histone peptides containing tert-butylnorleucine, tBuNle, a neutral isostere of Kme3. By comparing binding affinities for Kme3 versus tBuNle across systematically modified peptides and proteins, the team were able to quantify how much the aromatic cage contributes to overall binding, independent of interactions with surrounding histone residues.
The team examined three reader proteins representing different binding scenarios. For the CBX1 chromodomain, engineering mutations in the reader that strengthened interactions with residues flanking Kme3 reduced the protein's preference for charged Kme3 over neutral tBuNle. The SPIN1 Tudor domain showed a similar but smaller effect arising from stabilizing changes in the histone tail. In both of these cases, both overall cooperativity and the contribution of binding to Kme3 were reduced when interactions to the surrounding sequence were strengthened. In contrast, the SGF29 Tudor domain actually prefers tBuNle over Kme3, and the team traced this preference partly to anticooperativity: binding induces an unfavorable charge-charge repulsion between Kme3 and a nearby arginine on the histone tail that is relieved by replacement of the Kme3 with tBuNle.
These findings reveal that the contribution of interaction between Kme3 and the aromatic cage is influenced by cooperative interplay of multiple binding interactions across the protein-peptide interface. Critically, each reader displays a distinct degree of cooperativity. This variation offers a potential path toward selective inhibitors. Rather than targeting the conserved aromatic cage directly, drug designers might exploit differences in how strongly each reader couples PTM recognition to peripheral interactions. The work also demonstrates that mutations to either histones or readers could substantially alter PTM selectivity, with implications for understanding disease-associated variants. By establishing tBuNle as a quantitative probe for binding cooperativity, the Waters laboratory has provided the field with both mechanistic insight and a practical tool for therapeutic development against this challenging target class.