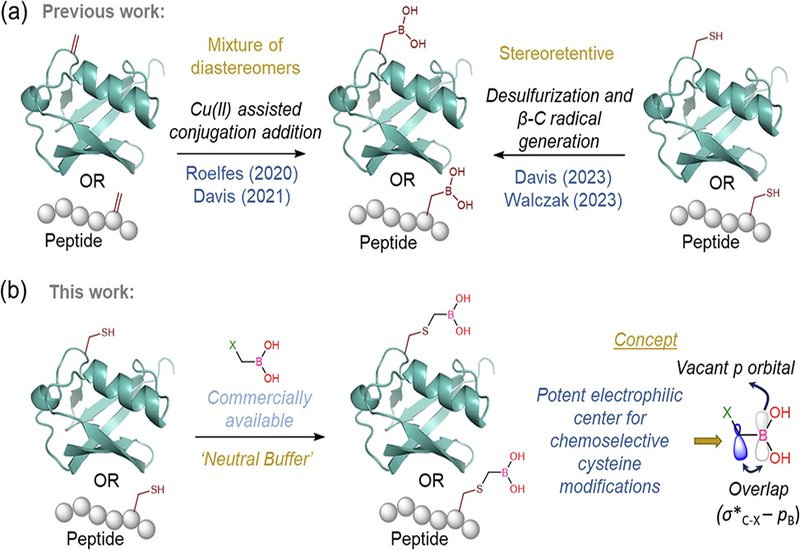
Boronic acids have earned a versatile reputation in chemical biology and drug design. Their empty p orbital allows dynamic switching between sp2 and sp3 hybridization, enabling reversible covalent capture of diols and making them excellent warheads for enzyme inhibitors, bioisosteres for carboxylic acids, and supramolecular recognition elements. Installing boronic acids onto peptides and proteins could unlock new therapeutic and diagnostic applications, yet existing methods face significant hurdles. Genetic code expansion suffers from boron's inhibitory effect on protein translation. Late-stage chemical approaches either require transition metals or photochemistry that cause unwanted side reactions, or they proceed through dehydroalanine intermediates that sacrifice stereochemistry. A simpler, cleaner strategy has remained elusive.
A team of researchers from the Indian Institute of Technology Ropar, supervised by Professor Anupam Bandyopadhyay, published in Nature Communications, hypothesized that commercially available halomethyl boronic esters might serve as ideal electrophiles for cysteine alkylation. DFT calculations revealed that the empty p orbital on boron overlaps with the σ* antibonding orbital of the adjacent carbon-halogen bond, activating the methylene center toward nucleophilic attack. Bromomethyl boronic acid pinacol ester emerged as the reagent of choice: reactive enough for rapid conversion yet mild enough to avoid over-alkylation. On the model peptide leucine enkephalin, the reaction achieved greater than 95% conversion within five minutes at physiological pH, with second-order rate constants exceeding 102 M−1s−1. The pinacol protecting group hydrolyzed spontaneously during HPLC purification, delivering free boronic acid handles without an extra deprotection step.
The method proved remarkably versatile. Eleven bioactive peptide sequences ranging from 4 to 36 residues underwent clean monoboronation at cysteine with isolated yields of 33 to 60 percent. Disulfide-containing peptides such as lanreotide required brief TCEP reduction before treatment, but then accepted two boronic acid units efficiently. The researchers extended this to tri- and tetravalent conjugates by designing sequences with multiple cysteine residues. Protein substrates followed suit: a 51-residue PDL1 construct, lysozyme, and bovine serum albumin all accepted boronic acid handles at their free thiols without perturbing native folds, as confirmed by circular dichroism. For BSA, subsequent labeling with a salicylhydroxamic acid fluorophore demonstrated that the installed boronic acid remained accessible for bioorthogonal conjugation.
The team then deployed their methodology to augment the imaging performance of the antimicrobial peptide UBI(29-41), a cationic sequence used clinically to visualize bacterial infections. They replaced two residues with cysteine-linked methylene boronic acids, creating a bivalent probe. The rationale: boronic acids should form reversible covalent bonds with the diol-rich glycans of wall teichoic acid and lipoteichoic acid on Staphylococcus aureus surfaces, synergizing with UBI's native electrostatic attraction. Flow cytometry confirmed a 40-fold improvement in staining intensity for the bis-boronated peptide compared to unmodified UBI. Confocal microscopy visualized S. aureus at concentrations as low as 50 nM without washing. The probe showed negligible binding to gram-negative Escherichia coli or mammalian cells, and its serum half-life increased 14-fold over native UBI. These results establish cysteine boronation as a practical, metal-free platform for installing boronic acid handles onto peptides and proteins, with immediate applications in precision bacterial imaging.