Tumor blood vessels differ from healthy vasculature in ways that drug developers can exploit. Endothelial cells lining tumor vessels overexpress E-selectin, a glycoprotein normally involved in immune cell trafficking, particularly at sites of active angiogenesis and metastasis. This localized overexpression creates an opportunity: nanocarriers bearing E-selectin-targeting ligands could concentrate at tumor sites while sparing healthy tissue. The natural ligand, Lewis X tetrasaccharide, binds E-selectin only weakly, but glycomimetic peptides such as IELLQAR and its cysteine-terminated variants CIELLQAR and CIELFQAR recognize the receptor with micromolar affinity, making them attractive targeting elements for drug delivery platforms.
A team led by Joel B. Alderete and Verónica A. Jiménez from Universidad de Talca and Universidad Andres Bello, Chile, respectively, publishing in Bioconjugate Chemistry, recognized that PAMAM dendrimers could provide an ideal scaffold for such targeting peptides. These branched polymers offer precise control over surface chemistry, established drug encapsulation properties, and proven biocompatibility when their charged surface amines are partially neutralized. However, acetylation that reduces toxicity also impairs cell penetration. The researchers reasoned that combining an E-selectin-targeting peptide with the cell-penetrating peptide pTAT on the same dendrimer could solve both problems: E-selectin recognition would direct the carrier to tumor vasculature, while pTAT would facilitate uptake once the carrier arrived.
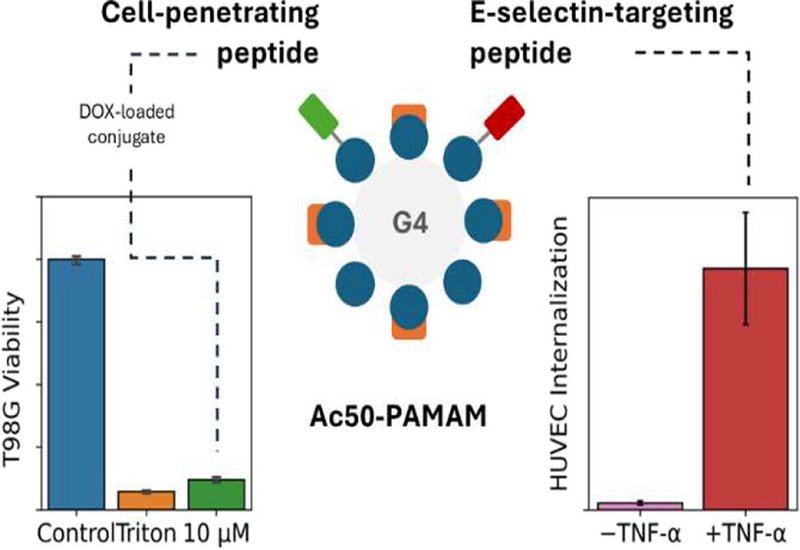
Fourth-generation PAMAM dendrimers were acetylated to approximately 50% surface coverage, then sequentially conjugated with pTAT and either CIELLQAR or CIELFQAR using a maleimide linker that couples to the peptides' N-terminal cysteines. NMR analysis confirmed attachment of 3–4 pTAT units and 2–3 E-selectin-targeting peptides per dendrimer. Both dual-peptide conjugates showed excellent safety profiles, maintaining greater than 80% cell viability across concentrations up to 10 μM in both endothelial and glioblastoma cell models after 24 and 48 hours of exposure.
The critical test came in endothelial cells activated with tumor necrosis factor-alpha to overexpress E-selectin, mimicking tumor vasculature conditions. After 12 hours of incubation, both conjugates entered activated cells, but their behavior diverged strikingly. The CIELFQAR-based conjugate showed marked preference for E-selectin-overexpressing cells compared to non-activated controls, exactly as designed. When cells were pre-treated with an anti-E-selectin antibody to block the receptor, uptake of the CIELFQAR conjugate was abolished, confirming E-selectin-mediated internalization. The CIELLQAR conjugate, by contrast, entered cells regardless of E-selectin status or antibody blocking, suggesting that its weaker receptor affinity allowed pTAT-mediated uptake to dominate. The 2-fold difference in binding affinity between the peptides, 16 μM for CIELFQAR versus 35 μM for CIELLQAR, apparently tips the balance between targeted and non-specific internalization.
The superior CIELFQAR conjugate also penetrated T98G glioblastoma cells, demonstrating that pTAT retains its cell-penetrating function in the dual-peptide format. Preliminary drug delivery experiments showed the conjugate could encapsulate doxorubicin and deliver it effectively: at 10 μM conjugate concentration, cell viability dropped to 60% after 24 hours and below 5% after 48 hours, matching the death control. The delayed cytotoxicity aligns with the observed 12-hour internalization requirement, suggesting the conjugate must first enter cells before releasing its cargo. By combining E-selectin targeting for vascular specificity with pTAT for cellular penetration, this dual-peptide dendrimer platform offers a promising architecture for tumor-directed drug delivery that exploits the distinctive molecular signature of pathological blood vessels.