The proto-oncogene MYC is dysregulated in more than 70% of human cancers and drives some of the most aggressive subtypes of breast cancer. Triple-negative breast cancer, which accounts for 10–15% of all breast cancer diagnoses, is strongly linked to MYC amplification and remains exceptionally difficult to treat. The health equity dimension is stark: 97% of breast tumors from African-American women show Myc overexpression, contributing to a 38% higher mortality rate compared to white women, and incidence among women in their 20s is rising. Myc exerts its oncogenic effects by partnering with transcription factor Max to bind the E-box DNA response element (5′-CACGTG), driving expression of roughly 15% of the genome. For decades, the extended dimerization interface of the Myc/Max/E-box network has resisted every small-molecule drug candidate tested against it, earning the label "undruggable."
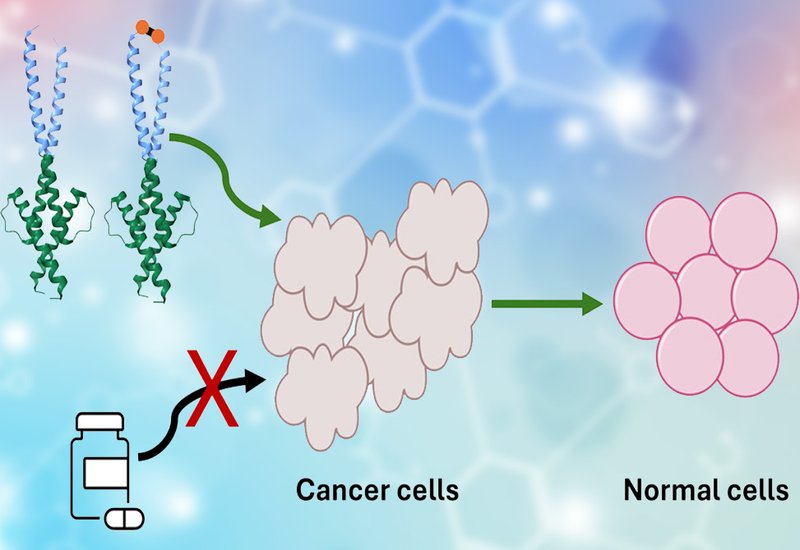
Researchers in the Shin Group at the University of Toronto, published in J. Med. Chem., engineered two frankenproteins, MEF and MEF/C93, by stitching together modules drawn from the basic region/helix–loop–helix/leucine zipper, bHLHZ, domains of transcription factors Max and Myc. Frankenproteins, as developed in the Shin lab, are chimeric proteins whose components are recruited from unrelated protein families and assembled into a single functional construct. The team built on their earlier protein ME47, which inhibited breast cancer xenograft growth in mice by more than 60%, and on MEF, which combined an evolved ME47 bHLH domain with the designed FosW leucine zipper for improved stability and E-box affinity. MEF/C93 extends MEF with a cysteine residue at the C-terminus of its FosW leucine zipper, enabling disulfide-linked dimerization under oxidizing conditions — conditions prevalent in both the bloodstream and in cancer cells. A built-in nuclear localization signal directs both proteins to the nucleus, where they compete with native Myc/Max for E-box occupancy.
Both MEF and MEF/C93 bound the E-box with dissociation constants near 8 nM in bacterial one-hybrid and quantitative electrophoretic mobility shift assays, rivaling native transcription factors in affinity and sequence specificity. Circular dichroism established that MEF/C93 achieves 91% α-helicity compared to 75% for MEF, a direct consequence of forced covalent leucine zipper dimerization. This structural enhancement carries pharmacological weight: stably folded, highly helical proteins resist protease degradation at the disordered regions that enzymes typically target, suggesting improved bloodstream lifetime for MEF/C93. Immunofluorescence confirmed that both proteins localized to the nuclei of MDA-MB-231 triple-negative breast cancer cells within 24 hours of treatment. Cell viability assays then delivered the central result: MEF and MEF/C93 exhibited IC50 values of 1–2 μM in Myc-dependent MDA-MB-231 cells, compared to approximately 25 μM in Myc-independent MCF-7 cells — a selectivity window of roughly 12-fold. Quantitative PCR reinforced the mechanism of action: both proteins selectively suppressed three canonical Myc target genes — PD-L1, nucleolin, and telomerase reverse transcriptase (TERT) — in MDA-MB-231 cells, with no measurable effect in MCF-7 cells, confirming that MEF and MEF/C93 act as competitive inhibitors of Myc/Max binding at the E-box specifically in Myc-driven cancer cells.
These results advance MEF and MEF/C93 as among the most selective protein-based inhibitors of the Myc/Max/E-box network reported to date and open a protein drug pipeline against a target that has defeated small-molecule approaches for decades. The study also carries a compelling health equity rationale: therapies that selectively neutralize Myc-driven tumors could provide outsized benefit to African-American women, who bear a disproportionate burden of MYC-amplified breast cancer and its associated mortality. The Shin Group plans to expand testing across additional Myc-dependent cancer cell lines and to evaluate pharmacokinetics and immunogenicity in mouse models, the next critical hurdles on the path from bench to clinic.