Alzheimer's disease is defined in part by the accumulation of amyloid beta, Aβ, peptides that resist normal clearance, aggregate into insoluble plaques, and drive a cascade of neuroinflammation, oxidative stress, and synaptic dysfunction. The central challenge in targeting this process therapeutically is that amyloid aggregates are largely inaccessible to conventional small molecules and antibodies, and the cellular pathways capable of clearing them, particularly the autophagy–lysosome system, have until recently lacked practical molecular handles for therapeutic intervention. Strategies such as PROTACs and LYTACs have begun to address this gap through the proteasomal and lysosomal routes respectively, but no natural product-derived peptide had yet been shown to act as a lysosome-targeting degrader capable of reducing Aβ-induced neurotoxicity.
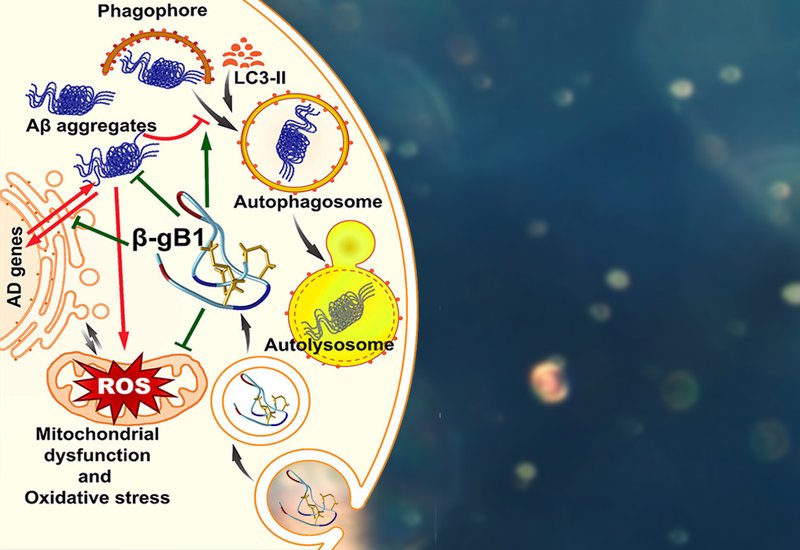
Researchers in the Tam Group at Nanyang Technological University identified a hyperdisulfide-constrained peptide from Ginkgo biloba nuts, termed β-ginkgotide β-gB1, as a first-in-class natural product-derived targeted protein degrader, published in Biochemistry. The 20-residue peptide is cross-braced by three disulfide bonds in a novel C–CC–C–CC cysteine spacing pattern not previously observed in plant peptides, yielding a cysteine content exceeding 30% and conferring exceptional resistance to thermal and proteolytic degradation. Crucially, β-gB1 harbors a canonical LC3-interacting region, LIR, motif within its central loop, a hexapeptide sequence that engages the autophagy machinery to promote selective degradation of intracellular cargo. The peptide was produced by Fmoc-based solid-phase synthesis followed by oxidative folding under mild aqueous conditions, and its identity was confirmed by co-elution with native β-gB1 on reverse-phase UHPLC.
Cellular studies in SH-SY5Y human neuroblastoma cells established that β-gB1 enters cells primarily through clathrin-mediated, energy-dependent endocytosis, distributing throughout the cytoplasm without nuclear accumulation. Pretreatment with 10 μM β-gB1 significantly protected cells against toxicity induced by both Aβ25–35 and Aβ1–42, as measured by MTT and lactate dehydrogenase assays, with an EC50 of 6.4 μM against Aβ25–35-induced neurotoxicity. Thioflavin T staining showed that β-gB1 co-treatment reduced Aβ aggregate formation by approximately 2.3- to 2.4-fold relative to Aβ-treated controls, while DCFH-DA fluorometry confirmed a corresponding reduction in reactive oxygen species. Autophagic vacuole staining with monodansylcadaverine revealed a 4-fold increase in autophagy activity in β-gB1 co-treated cells, alongside a 1.6-fold reduction in propidium iodide-positive cell death compared to Aβ-treated groups alone.
Gene expression analysis by qRT-PCR further demonstrated that β-gB1 co-treatment normalized the Aβ-induced dysregulation of a broad panel of AD-related genes, suppressing overexpression of APP, MAPT, GSK3B, ACHE, and CD33, while restoring the expression of genes associated with Aβ homeostasis and clearance, including APOE, LRP1, ADAM10, and the presenilins. Taken together, the structural stability, cell-penetrating capacity, LIR-mediated autophagy induction, and demonstrated neuroprotective activity of β-gB1 position it as a promising lead compound and scaffold for developing intracellularly active therapeutics targeting amyloid-driven neurodegeneration.