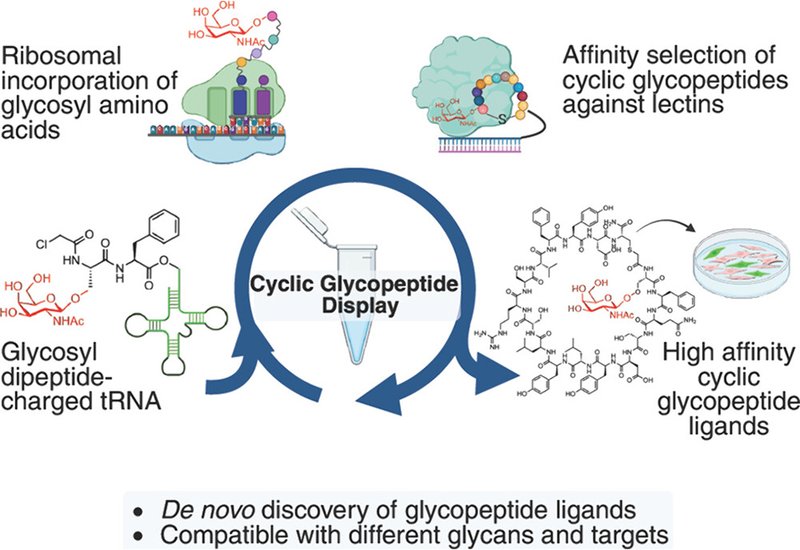
Macrocyclic peptides have emerged as powerful molecular tools for targeting protein interactions, particularly those inaccessible to small molecules. Display technologies such as mRNA display enable de novo discovery from libraries exceeding 1012 sequences, and when coupled with flexizyme-mediated genetic code reprogramming, researchers can incorporate non-natural amino acids to enhance binding and drug-like properties. Yet one major modification has remained out of reach: glycosylation. Protein glycosylation drives high-affinity interactions with lectins, a family of carbohydrate-binding proteins increasingly recognized as drug targets and receptors for targeted protein degradation. Despite intense interest, de novo selection of glycopeptide ligands bearing native glycosidic linkages has not been reported, largely because ribosomes fail to efficiently incorporate bulky glycosyl-amino acids during translation.
Research in the Payne Group at the University of Sydney, published in the Journal of the American Chemical Society, have now overcome this barrier with a dipeptide reprogramming strategy that enables ribosomal incorporation of glycosyl-amino acids into cyclic peptides displayed on mRNA. The team initially attempted to reprogram the initiator position with ClAc-L-Ser(α-L-Fuc)-DBE, reasoning that L-fucose, a natural post-translational modification critical for selectin binding, would be a valuable starting point. Flexizyme-mediated aminoacylation of initiator tRNA succeeded, but in vitro translation failed: the major product arose from deletion of the first two residues, indicating that the bulky sugar clashed sterically within the ribosome P-site. The solution was to move the glycosyl-amino acid further into the exit tunnel by initiating translation with a dipeptide. Using synthetic ClAc-L-Ser(α-L-Fuc)-L-Phe-CME as a substrate for the eFx flexizyme, the group achieved clean formation of a model macrocyclic glycosulfopeptide bearing both α-fucosyl-serine and sulfotyrosine, validating compatibility with dual genetic reprogramming.
The team applied the dipeptide strategy to mRNA display selection against P-selectin, an adhesion molecule on activated platelets that mediates leukocyte-platelet aggregation via its ligand PSGL-1. A semi-randomized DNA library encoding more than 1012 unique sequences was transcribed, ligated with puromycin, and translated with reprogramming of both N-ClAc-L-Ser(α-L-Fuc)-L-Phe at the initiator position and H-L-Tyr(SO3−) at internal AUG codons. After six rounds of panning against immobilized His6-tagged P-selectin, sequencing revealed distinct peptide families. Eight top sequences were synthesized by Fmoc solid-phase peptide synthesis, and four exhibited moderate to high affinity for P-selectin by surface plasmon resonance, with KD values ranging from 1.1 to 8.2 μM. The highest-affinity peptide, P6, matched the reported affinity of recombinant PSGL-1 for P-selectin at approximately 0.8 μM. Critically, des-fucosylated analogues of three peptides showed dramatic losses in binding, confirming that the glycan drives affinity. In functional assays, peptides P3, P5, and P6 suppressed platelet-induced monocyte aggregate formation in human blood by more than 50% at 20 μM, demonstrating potent disruption of P-selectin-mediated platelet-leukocyte interactions under prothrombotic conditions.
The group next targeted the asialoglycoprotein receptor, ASGPR, a C-type lectin on hepatocytes that internalizes glycoproteins bearing terminal galactose or N-acetylgalactosamine, GalNAc, residues and delivers them to lysosomes. ASGPR is the receptor exploited by lysosome-targeting chimeras, LYTACs, in which tri-GalNAc drives extracellular protein degradation. The researchers synthesized ClAc-L-Ser(β-D-GalNAc)-L-Phe-CME and confirmed efficient incorporation into a translated model peptide. A library reprogrammed with L-Ser(β-D-GalNAc) was panned against biotinylated ASGPR for seven rounds, and the top ten enriched sequences were synthesized. Peptide A9 exhibited a KD of 410 nM by SPR, while the des-GalNAc control, des-A9, showed negligible binding with KD exceeding 100 μM, underscoring the essential role of the carbohydrate. In a fluorescence polarization assay, A9 competed with fluorescently labeled tri-GalNAc for ASGPR binding with an IC50 of 0.58 μM, approximately 24-fold more potent than free GalNAc. The team prepared bifunctional biotin(PEG8)A9 conjugates and demonstrated dose-dependent uptake of fluorescent streptavidin into HepG2 liver cells by flow cytometry, with efficiency comparable to biotin(PEG8)GN3. Confocal microscopy confirmed colocalization of internalized streptavidin with the lysosomal marker LAMP1, demonstrating that monomeric cyclic glycopeptide A9 functions as a LYTAC warhead and traffics cargo to lysosomes.
This work provides the first general platform for ribosomal translation and mRNA display of glycopeptides bearing native glycosidic linkages. The dipeptide reprogramming strategy overcomes steric constraints that have long thwarted incorporation of bulky post-translational modifications and opens access to vast libraries of glycopeptide ligands for lectin targets. The P-selectin inhibitors and ASGPR agonists represent novel chemotypes for modulating platelet-leukocyte cross-talk and for next-generation targeted protein degraders, respectively. Although the current method is limited to N-terminal glycosylation, the technology is likely to extend to other large modifications and enable de novo discovery campaigns across a broad range of glycan-binding proteins.