α-Synuclein, αSyn, is a 140-residue presynaptic protein whose aggregation into amyloid fibrils lies at the heart of Parkinson's disease and related synucleinopathies. Although the central non-amyloid component, NAC, region spanning residues 65–90 forms the fibril core, mounting evidence points to two short N-terminal motifs, P1, αSyn[36–42], and P2, αSyn[45–57], as the gatekeepers of assembly. Deletion of P1 alone is sufficient to block fibrillation at neutral pH, and a single substitution, L38M, abolishes nucleation without preventing elongation from wild-type seeds. How a single methionine introduction at position 38 can so profoundly rewire the aggregation landscape has remained mechanistically obscure.
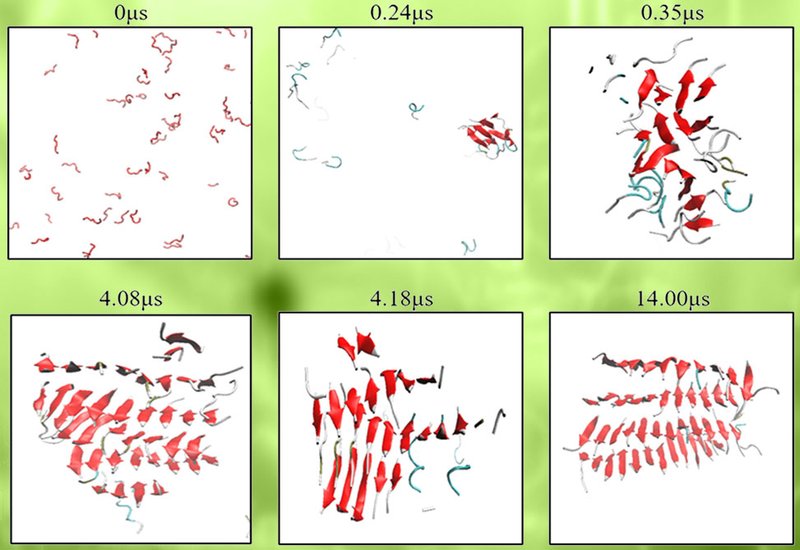
Researchers in the Hall Group at North Carolina State University, in collaboration with those in the Radford and Brockwell Groups at Leeds University, published in Protein Science, deployed discontinuous molecular dynamics simulations with the coarse-grained PRIME20 force field to answer that question. The team simulated spontaneous self-assembly of P1, P2, P3, αSyn[36–57], and the extended P3Next, αSyn[27–57], fragment in their wild-type forms alongside the L38M, L38A, and V40A substitutions and, as secondary variants, Y39A and S42A. Fifty-nine independent simulations were run in total, with control experiments on a low-amyloid-propensity fragment, C1ext, αSyn[14–35], confirming that PRIME20 does not systematically overpredict aggregation. Representative coarse-grained fibrils were subsequently reconstructed to all-atom resolution and validated via six independent 500-ns explicit-solvent trajectories.
Every wild-type fragment assembled into β-sheet-rich fibrillar structures, but with distinct architectures. P1-WT favored multilayer parallel β-sheets, while P2-WT, whose Gly51 introduces a natural kink, curved into U-shaped or S-shaped single-layer assemblies reminiscent of the experimentally characterized PreNAC nanocrystal. P3-WT produced polymorphic mixed-registry sheets, and P3Next-WT consistently nucleated β-hairpins, averaging 23% ± 1% hairpin content across seven replicas. This heterogeneity mirrors the structural polymorphism seen experimentally in full-length αSyn oligomers. The L38M substitution stood apart from all other variants tested. In P1-L38M fibrils, the fraction of antiparallel residues rose to 35% ± 2% compared with just 10% ± 3% in P1-WT, and in P3Next-L38M fibrils the β-hairpin content fell to 16% ± 2%, significantly lower than the 23% ± 1% of P3Next-WT, p = 0.009. Crucially, structural analysis revealed that residue 38 in L38M variants preferentially occupies a longer, four-residue loop, whereas in WT, L38A, and V40A variants it sits at the strand-loop boundary within a tighter three-residue turn. Methionine's bulkier side chain appears to demand extra loop space, destabilizing the very hairpin geometry that normally primes oligomers for fibril conversion. By contrast, L38A and V40A maintained or increased hairpin content relative to WT, consistent with their negligible effect on full-length αSyn fibrillation observed experimentally.
From these results the team proposed two mechanistic pathways by which L38M suppresses full-length αSyn amyloid formation. The first invokes reduced β-hairpin formation in the P1/P2 region, depriving early oligomers of the intermolecular binding elements thought to drive oligomer-to-fibril conversion. The second invokes the accumulation of antiparallel oligomers that are either kinetically trapped off-pathway or must rearrange into parallel architecture before fibril growth can proceed, a costly reorganization that slows or stalls the process entirely. Both pathways are consistent with prior experimental reports linking antiparallel β-sheet content to prefibrillar, non-amyloid-competent species. The simulations reframe L38M not as a sequence intrinsically averse to aggregation, but as one that subtly corrupts the supramolecular geometry needed to convert oligomers into propagating fibrils. Taken together, the findings establish the P1/P2 β-hairpin as a druggable structural checkpoint in αSyn amyloidogenesis and suggest that small molecules or peptide mimetics capable of lengthening or otherwise destabilizing the P1 loop could replicate the protective effect of L38M in a therapeutic context.