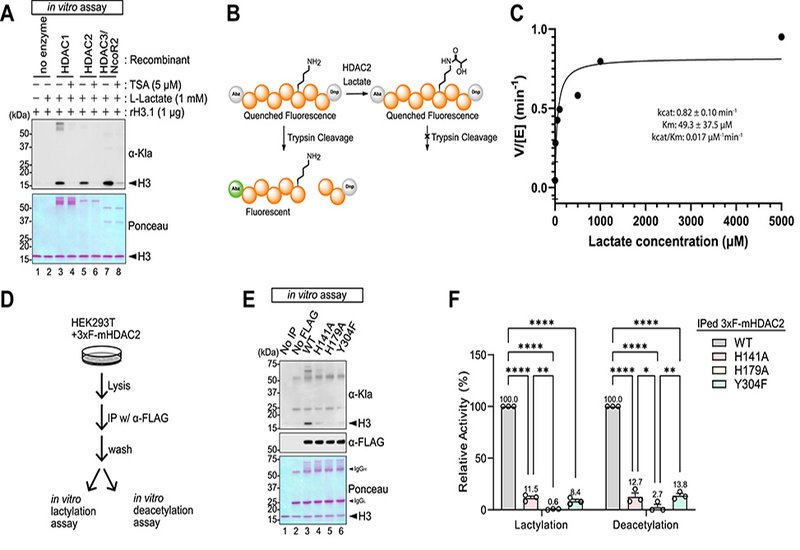
Lysine lactylation, Kla, has emerged as a post-translational modification, PTM, directly linking glycolytic metabolism to protein regulation. While earlier models invoked lactyl-CoA–dependent transfer or non-enzymatic reactions via lactoylglutathione, the enzymatic basis of Kla remained uncertain. Building on evidence that class I histone deacetylases, HDACs 1–3, catalyze β-hydroxybutyrylation, a study published in the Journal of Biological Chemistry by researchers in the Burslem and Goldberg Labs at the University of Pennsylvania and University of California, San Francisco, respectively, shows that the same enzymes also directly catalyze lysine lactylation.
Using recombinant proteins, reconstitution assays, and LC–MS/MS, the authors confirmed that HDACs 1, 2, and 3 catalyze Kla on multiple histone lysines, with activity blocked by the pan-HDAC inhibitor trichostatin A. Kinetic analysis revealed parameters, kcat ≈ 0.82 min-1, Km ≈ 49 μM, consistent with physiological lactate concentrations, supporting a model in which HDACs use ambient lactate directly, independent of lactyl-CoA. Dialysis experiments confirmed reversibility: HDACs add or remove lactate depending on substrate availability.
Cellular assays reinforced this catalytic role. Genetic depletion of HDAC1–3 in HEK293T cells reduced global Kla, with triple knockdown producing the strongest effect. Pharmacological inhibition decreased Kla without altering intracellular lactate or lactyl-CoA levels, uncoupling Kla from CoA dependence and pointing directly to HDAC activity. In LDHA/B double knockout cells, loss of glycolytic lactate production abolished Kla, but exogenous l-lactate rescued it, highlighting lactate itself as the substrate. In bone marrow–derived macrophages, LPS/IFNγ-induced glycolysis drove Kla accumulation that was blocked by HDAC inhibitors, showing relevance to immune activation. Glucose availability and inhibitors of lactate production, oxamate, dichloroacetate, modulated Kla, underscoring its tight dependence on metabolic flux.
Knockdown of acyltransferases, p300, CBP, HBO1, and lactyl-CoA synthetases, ACSS2, SUCLG2, had little effect on basal Kla, demonstrating that the dominant pathway in proliferating cells is HDAC-mediated. Other routes, such as AARS-dependent transfer or lactoylglutathione–derived d-lactylation, may contribute under specialized conditions but are not the primary drivers in standard settings.
These findings reposition class I HDACs as bifunctional acylation enzymes capable of both erasing and depositing short-chain acyl marks, expanding their role beyond canonical deacetylation. The results highlight how glycolysis-derived lactate, produced at millimolar levels, directly influences chromatin and protein regulation. The biological functions of Kla remain unresolved, but its induction in activated macrophages points to a role in inflammatory gene programs.
This study establishes class I HDACs as major enzymatic drivers of lysine lactylation, acting through a reversible, lactate-dependent condensation reaction. By decoupling Kla formation from lactyl-CoA and demonstrating its reliance on glycolytic flux, the work provides a mechanistic framework that directly connects metabolism, chromatin state, and PTM dynamics. Future efforts will need to resolve the targets, stoichiometry, and physiological consequences of HDAC-catalyzed Kla.