Hierarchical superstructures assembled from peptides and lipids underpin a wide range of biological functions, from membrane-active antimicrobial action to lipoprotein transport, and have attracted growing interest as platforms for biomimetic material design. A key but poorly understood variable in these assemblies is peptide secondary structure. While it is known that conformational state influences how peptides interact with lipid membranes, the thermodynamic and kinetic principles that govern which hierarchical architectures actually form, and by which pathway, have remained largely opaque. Existing experimental techniques lack the temporal and spatial resolution to capture intermediate states during multicomponent assembly, leaving the mechanistic picture incomplete.
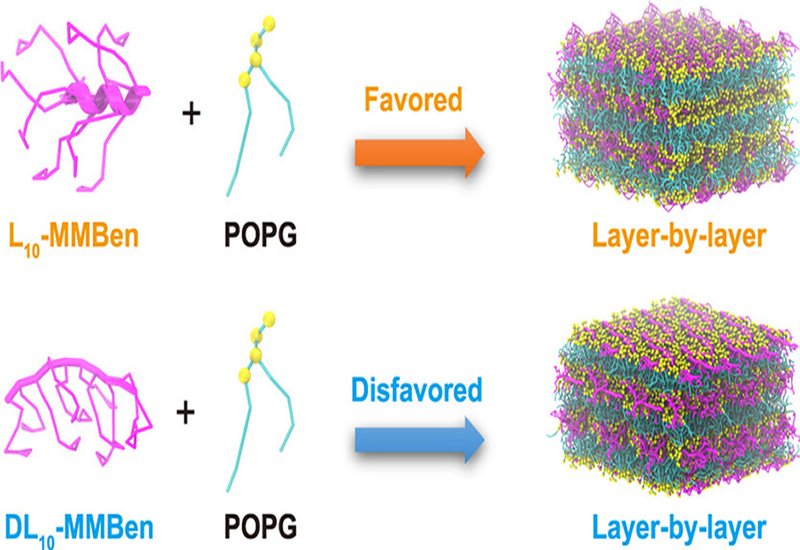
Researchers supervised by Professor Shiyan Xiao at the University of Science and Technology of China in Hefei, published in J. Am. Chem. Soc., studied the assembly of the phospholipid POPG with radially amphiphilic peptides, RAPs, using coarse-grained molecular dynamics, CGMD, simulations combined with Markov state models, MSMs. The RAP pair examined, L10-MMBen and its stereochemical counterpart DL10-MMBen, share identical primary sequences but differ in residue chirality: L10-MMBen adopts an α-helical conformation, while DL10-MMBen is unstructured. This chemically matched pair allowed the role of secondary structure alone to be isolated from sequence effects. Free-energy landscapes were computed for single-layered, double-layered, and triple-layered assembly systems, with over 200 simulation replicas per system to generate statistically robust pathway data.
The two RAPs drove POPG assembly along sharply divergent trajectories. In triple-layered systems, α-helical L10-MMBen produced defect-free layer-by-layer superstructures in 72.9% of replicas; unstructured DL10-MMBen reached the same endpoint in only 17.5%. Free-energy landscape analysis revealed two competing pathways from an initial disordered state: a singular-layer pathway terminating in single bilayers, and a layer-by-layer pathway yielding stacked multilayer architectures. Both RAPs pass through the same porous-rich layer intermediate, but their subsequent fates differ dramatically. For L10-MMBen, transfer into the layer-by-layer pathway proceeds at a rate 10 times faster than for DL10-MMBen at the initial assembly step, and the kinetic preference for the layer-by-layer pathway over the singular-layer pathway is strongly pronounced. For DL10-MMBen the reverse holds, with the singular-layer route proceeding roughly 150 times faster than the layer-by-layer route from the porous-rich layer state. Enthalpy-entropy decomposition identified the mechanistic basis for this divergence: the enthalpic barrier for transitioning from the porous-rich layer intermediate to the defect-free triple bilayer is 1.805 kJ/mol per lipid for L10-MMBen but 3.076 kJ/mol per lipid for DL10-MMBen. The helical conformation of L10-MMBen exposes a larger hydrophilic surface area, weakening its interactions with POPG and enhancing lateral diffusion of both the peptides and the lipid molecules. This increased fluidity enables the system to escape long-lived metastable states, including the porous-rich layer and cross-cylinder configurations. Potential of mean force calculations further showed that helical RAPs destabilize the cylindrical stalk structures that bridge membranes in these metastable states, with a stalk disruption free-energy change of 122.24 kJ/mol for L10-MMBen versus 107.37 kJ/mol for DL10-MMBen, actively promoting progression toward layered superstructures rather than stalling there.
These findings establish secondary structure as a programmable handle for controlling not just the final morphology of peptide-lipid assemblies but the kinetic pathway by which that morphology is reached. The work challenges the conventional framing of lipid self-assembly as driven purely by entropy or purely by enthalpy, demonstrating instead that both contributions vary dynamically across assembly stages. For biomaterial designers, the results suggest that engineering α-helical conformation into amphiphilic peptides offers a reliable strategy for promoting ordered, defect-free multilayer architectures, with implications for membrane-active therapeutics, drug delivery scaffolds, and the rational design of functional hierarchical biomaterials.