α-Helical protein-protein interactions govern countless biological processes, yet short peptides designed to mimic these interfaces rarely maintain stable helices in solution. α,α-Disubstituted α-amino acids, dαAAs, offer a solution: their gem-disubstituted backbone locks peptides into helical conformations while resisting proteolytic attack. However, incorporating dαAAs into high-throughput discovery platforms has proven difficult. Ribosomal translation yields poor efficiency, and chemical library methods cap diversity at roughly 108 members.
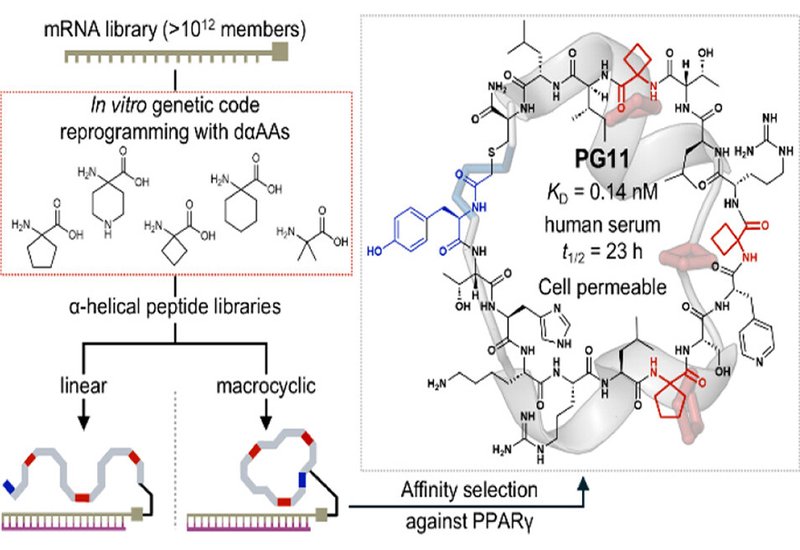
A team led by Hiroaki Suga and Takayuki Katoh at the University of Tokyo extended their engineered tRNA system, tRNAPro1E2, to incorporate five achiral dαAAs into nascent peptide chains via flexizyme-mediated charging. By tuning elongation factor concentrations, they achieved site-specific incorporation of up to five identical or four different dαAAs per peptide. Armed with this capability, they constructed linear and macrocyclic libraries exceeding 1012 members and screened them against PPARγ, a nuclear receptor whose coregulator recruitment depends on an α-helical surface known as AF-2.
RaPID selection delivered diverse hits with low-to-subnanomolar affinities; the most potent, PG11, bound PPARγ at 140 pM. X-ray crystallography at resolutions as fine as 1.4 Å revealed helix-turn-helix conformations docking into the AF-2 coregulator cleft. The dαAAs sat within helical segments but formed no direct protein contacts, consistent with a conformational preorganization role rather than direct recognition. Alanine substitution weakened binding up to 90-fold and accelerated proteolytic degradation dramatically: PG08 displayed a serum half-life of 100 hours versus 5.6 hours for its alanine mutant. Several peptides penetrated cells as efficiently as the benchmark R9 sequence and antagonized pioglitazone-induced PPARγ activation in Gal4 reporter assays, confirming functional intracellular target engagement.
By enabling ribosomal incorporation of multiple dαAAs into trillion-member libraries, this platform leapfrogs the diversity ceiling of chemical synthesis and sidesteps the incompatibility of phage and yeast display with non-natural backbones. The resulting peptides combine helical stability, protease resistance, and cell permeability without requiring stapling chemistry or cell-penetrating peptide fusions. As α-helical interfaces underpin interactions from oncogenic transcription factors to viral entry machinery, dαAA-based RaPID selection stands poised as a generalizable discovery engine for a historically challenging target class.