Transcription factors regulate thousands of genes and, when dysregulated, drive cancers ranging from melanoma to colorectal disease. Yet these proteins remain among the most difficult drug targets in medicine. Their flat, dynamic binding surfaces offer no pockets for conventional small molecules to grip, earning them the label "undruggable." Peptides can engage these expansive interfaces, and chemical cyclization can lock peptides into stable, bioactive conformations. However, discovering the right combination of peptide sequence and optimal cyclization geometry has required exhaustive trial and error, with researchers synthesizing hundreds of constrained variants after the fact to find productive stapling sites.
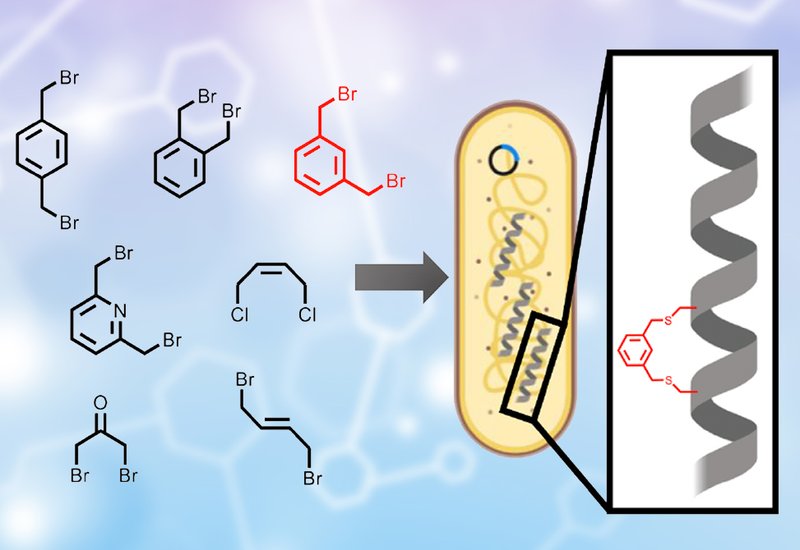
Researchers in the Mason Group at the University of Bath, published in Cell Chemical Biology, developed a platform called intracellular-cyclization TBS that merges chemical peptide stapling with live-cell functional screening in a single step. The team demonstrated that bis-alkylating reagents, small molecules bearing two reactive halide groups, can cross bacterial membranes and selectively bridge engineered cysteine pairs on peptides expressed inside E. coli. Integrated with their transcription block survival assay, this approach generates and tests millions of chemically constrained peptide variants simultaneously. Bacteria survive only if the stapled peptide they produce successfully blocks the target transcription factor from binding DNA, so the most potent inhibitors naturally dominate the culture over successive rounds of selection.
The team targeted CREB1, an oncogenic transcription factor overactive in melanoma, colorectal, prostate, and breast cancers. Screening libraries of over 63 million cyclic peptide variants, the platform converged on antagonists with nanomolar binding affinity. The lead CDCB-cyclized peptide bound CREB1 at 41 nM by isothermal titration calorimetry and disrupted the CREB1-DNA interaction with an IC50 of 247 nM, roughly twice as effective as the best linear counterpart. Critically, retroactive cyclization of the linear winner peptide at the same stapling position actually reduced its efficacy by over nine-fold, demonstrating that sequence and constraint geometry must evolve together. When conjugated to a cell-penetrating peptide derived from bovine lactoferricin, the lead compound entered A375 melanoma cells, suppressed CREB1-dependent transcription, reduced levels of the oncogenic proteins KPNA2 and ACOT7, and triggered apoptosis. The peptide also reduced HCT116 colorectal cancer cell viability at comparable potencies while showing minimal toxicity in non-cancerous fibroblasts.
This work establishes that post-translational chemistry and genotype-to-phenotype selection can operate in tandem inside living cells, accessing structural diversity beyond what genetic encoding alone permits. Built on standard bacterial genetics and inexpensive media additives, the platform offers a scalable and sustainable route to constrained peptide therapeutics against protein-DNA interfaces that have long resisted conventional drug discovery.