Discovering potent peptide ligands against proteins with shallow binding pockets, large protein-protein interaction surfaces, or no obvious druggable groove remains a central challenge in chemical biology. One well-established strategy to raise affinity is to link two independent binders so they engage a target simultaneously, but this approach traditionally requires pre-existing monomer ligands and structural knowledge of the target. A platform that could identify complementary peptide pairs de novo from randomized libraries, without any prior structural information, would expand the scope of macrocyclic peptide drug discovery considerably.
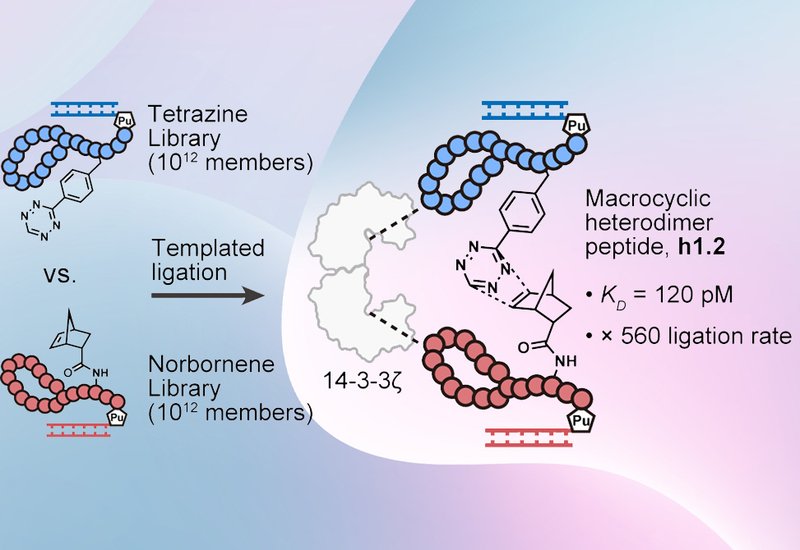
Researchers in the Vinogradov Group at the National University of Singapore and the Suga Group at The University of Tokyo, published in the Journal of the American Chemical Society, developed a library-vs-library selection platform that pairs two independently displayed macrocyclic peptide libraries in the presence of a target protein. The platform, Random nonstandard Peptides Integrated Discovery, RaPID, uses genetic code reprogramming to incorporate nonproteinogenic amino acids bearing mutually reactive groups, a tetrazine in library A and a norbornene in library B, into ribosomal peptides each encoded by its own mRNA. When two peptides from opposite libraries co-bind one protein molecule, their reactive handles are brought into proximity and undergo an inverse electron demand Diels-Alder, IEDDA, reaction to forge a covalent heterodimer. Tandem affinity purification with orthogonal HA and biotin tags, followed by separation PCR, then selectively recovers only the ligated conjugate and its encoding nucleic acids.
The team first validated tandem purification using the known coiled-coil pair E3 and K3, confirming selective recovery of covalently linked conjugates with up to 600-fold enrichment over controls. Moving to the therapeutic target 14-3-3ζ, a homodimeric adaptor protein implicated in cancer and neurodegeneration, an initial attempt with slower 1,4-addition chemistry between cysteine and acrylamide failed to yield covalent heterodimers. LC/MS analysis and heat-denaturation experiments showed that recovery in those rounds arose from noncovalent ternary complexes, not covalent ligation. Replacing the warheads with an IEDDA reactive pair resolved this problem: the Dab(H-Tz)/Dab(Norb) combination reacted up to 600-fold faster than the acrylamide/cysteine system, and adding a 70 °C denaturation step before tandem purification eliminated noncovalent background. Five rounds of IEDDA selection converged on three conserved sequence motifs across both libraries.
To identify which library A and library B sequences formed productive heterodimers, the team conducted reselections pairing individual hit sequences from library B against the full fifth-round library A pool, scoring pairs with a custom H-score metric. The analysis pointed to the a1/b2 pair. Chemically synthesized a1 and b2 incubated with 14-3-3ζ formed the heterodimer h1.2 cleanly by LC/MS, and the protein accelerated the initial rate of ligation more than 560-fold. Surface plasmon resonance revealed that h1.2 bound 14-3-3ζ with a KD of 120 ± 50 pM, compared with 430 ± 70 pM and 700 ± 300 pM for monomers a1 and b2, respectively. The affinity gain of h1.2 over the monomers arose primarily from a dissociation rate constant more than four-fold slower than those of the individual peptides. By contrast, the known bivalent ligand difopein, a double repeat of the phage-display-derived peptide R18, showed no improvement in binding affinity relative to the R18 monomer, illustrating that simple dimerization without linker optimization does not reliably enhance potency.
The library-vs-library RaPID platform addresses a gap that fragment-based and kinetic target-guided synthesis methods leave open: the library sizes accessible by mass spectrometry decoding rarely exceed a few hundred compounds, and structural knowledge of the target is typically required to design reactive fragment pairs. By encoding both reactive handles and variable-length linkers directly in the mRNA sequence, the present approach simultaneously optimizes pharmacophore identity and inter-fragment geometry across a search space of roughly 1012 × 1012 combinations. The authors note that the promiscuous pairings observed for 14-3-3ζ likely reflect the homodimeric architecture of the target, where each cyclic peptide monomer engages an equivalent subunit, and expect the strategy to perform with greater selectivity against monomeric or multidomain proteins. Future directions include applying the platform to targets with featureless surfaces, as well as extending the chemistry toward molecular-glue-like bivalent heterodimers and receptor dimerization modulators.