Fluorine is among the most consequential substituents in medicinal chemistry, improving metabolic stability, membrane permeability, and target binding across a broad range of drug candidates. An estimated twenty percent of all approved pharmaceuticals contain at least one fluorine atom. Yet delivering fluoride nucleophilically with both efficiency and stereochemical control remains a formidable synthetic challenge. Alkali metal fluorides such as cesium fluoride, CsF, and potassium fluoride, KF, are the safest and most economical fluorine sources available, but their near-total insolubility in organic solvents severely limits their utility in asymmetric catalysis. The enzyme fluorinase, which mediates the only known biological carbon-fluorine bond-forming reaction, solves this problem through a precisely tuned hydrogen-bonding network in its active site that desolvates and activates fluoride for nucleophilic substitution. Drawing on this enzymatic blueprint, researchers in the Gouverneur Group at the University of Oxford developed peptide-based catalysts for asymmetric nucleophilic fluorination with CsF, publishing their findings in the Journal of the American Chemical Society.
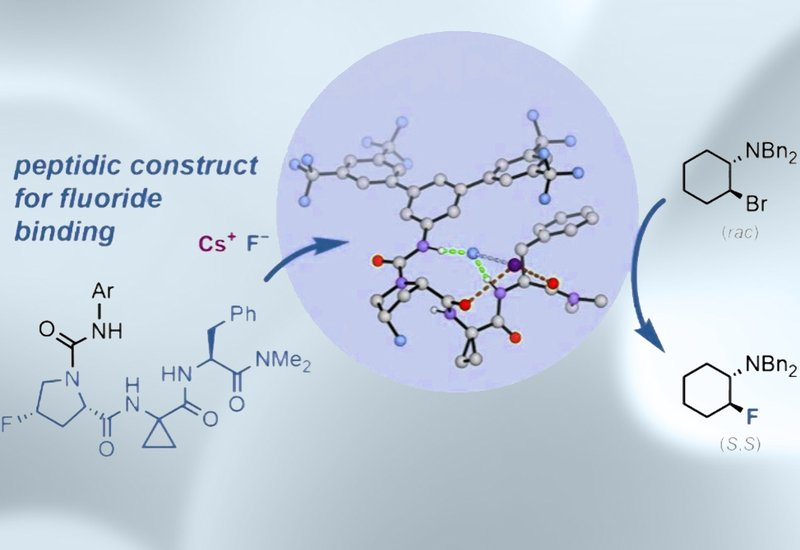
Although halide-peptide interactions have been studied extensively for chloride and other anions, peptide-fluoride binding had previously been explored only by computational methods. To fill this gap, the team designed a series of short tetrapeptides incorporating a proline, Pro, and 1-aminocyclopropane-1-carboxylic acid, Acpc, motif known to enforce a β-turn geometry. This conformational constraint was intended to position backbone amide NH hydrogen-bond donors, HBDs, around a central binding cavity suited for fluoride chelation. A lead tetrapeptide was titrated with tetrabutylammonium fluoride trihydrate, TBAF·3H2O, in deuterated dichloromethane, revealing 1:1 binding with moderate affinity. NMR chemical shift analysis showed that only a single NH group engaged fluoride, indicating monodentate binding. To shift toward multidentate coordination, the team introduced an electron-deficient N-terminal urea cap bearing trifluoromethyl-substituted aryl groups, which disrupted competing intramolecular hydrogen bonds and freed additional NH donors for fluoride chelation. This modification converted binding from mono- to tridentate and increased association constants by two orders of magnitude. Further conformational tuning through replacement of l-proline with the conformationally locked, 4S-fluoroproline, flp, stabilized the β-turn through a Cγ-endo ring pucker and raised affinities to association constants, Ka, of up to 4.0 × 104 M-1 for TBAF·3H2O.
With the optimized peptide catalyst in hand, the team demonstrated the first peptide-catalyzed asymmetric nucleophilic fluorination reactions. Using CsF as the fluoride source and racemic β-haloamine substrates that ionize to reactive meso-aziridinium intermediates under reaction conditions, the best-performing catalyst delivered fluorinated products in up to 86% yield and 88:12 enantiomeric ratio, e.r., at 10 mol% loading. Gram-scale fluorination retained performance at 83% yield and 89:11 e.r. at 5 mol% loading. The substrate scope produced precursors to several bioactive compounds, including a β-secretase, BACE, inhibitor, adenosine A1 receptor agonists, a galectin-1 and galectin-3 inhibitor, and a coagulation factor Xa inhibitor. NMR studies combined with Global Optimization Algorithm, GOAT, density functional theory, DFT, calculations revealed a mechanistic distinction between CsF and TBAF binding: whereas TBAF promoted tridentate fluoride coordination, CsF produced bidentate fluoride binding, with the cesium cation simultaneously chelated by two peptidic carbonyl groups. This dual chelation mode, engaging both the fluoride anion through NH donors and the cesium cation through carbonyls, accounts for the ability of the peptide to solubilize and activate CsF as a phase-transfer agent.
These results establish peptides as a previously unrecognized scaffold class for fluoride binding and catalytic fluorination, and provide the first experimental framework for understanding how peptide conformation governs fluoride chelation. The modularity of the peptide backbone, combined with the accessibility of non-canonical residues such as fluorinated prolines and electron-withdrawing urea caps, defines a tunable platform for expanding the scope of asymmetric fluorination chemistry. The authors note that further optimization through machine learning-assisted screening and expanded chemical space exploration may yield peptidic catalysts with substantially enhanced enantioselectivity and substrate generality, and potentially open a route toward peptide-based fluorination enzymes.