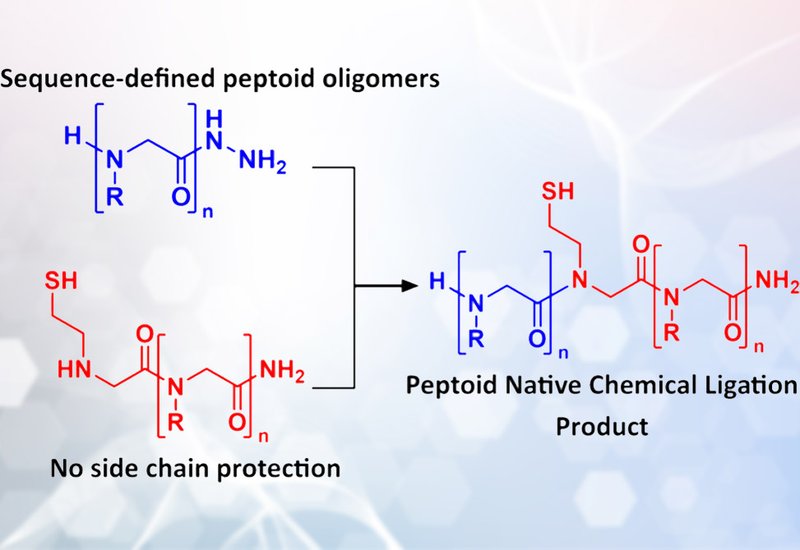
Peptide chemists have long used native chemical ligation, NCL, to overcome a fundamental limitation: the longer the chain, the lower the yield. By joining shorter, high-yielding fragments into full-length proteins, NCL enables ambitious syntheses that would otherwise fail. The strategy relies on selective reaction between a C-terminal thioester and an N-terminal cysteine, producing a native amide bond without protecting groups. Peptoid chemists have envied this capability for years, but a critical obstacle has blocked progress. Peptoid synthesis uses primary amines as building blocks under basic conditions, incompatible with the acid-labile thioesters required for NCL. Without fragment condensation methods, peptoid chemists face a stark trade-off between sequence control and chain length.
Researchers in the Kirshenbaum Group at New York University, published in Biochemistry, adapted the hydrazide ligation technique recently developed for peptides to enable native chemical ligation of peptoid oligomers. The team synthesized peptoid fragments bearing C-terminal hydrazides using standard solid-phase submonomer chemistry, initiating synthesis from hydrazine resin with an Fmoc-protected amino acid to circumvent the incompatibility of bromoacetic acid coupling. Partner fragments incorporated an N-terminal cysteamine-derived side chain to provide the thiol nucleophile. Oxidation of the hydrazide with sodium nitrite at pH 3 generated an acyl azide intermediate, which was converted to a thioester in situ using 4-mercaptophenylacetic acid. Subsequent ligation at neutral pH proceeded through the canonical five-membered ring intermediate, with the S-to-N acyl shift mediated by the secondary amine backbone characteristic of peptoids.
Model oligomer ligations demonstrated broad tolerance for side chain diversity. Short peptoid hexamers and heptamers incorporating aromatic, aliphatic, and charged side chains ligated efficiently, with some conversions approaching 90 percent. The protocol accommodated variation in both the C-terminal residue, with N-benzylglycine substituting for sarcosine, and the N-terminal thiol-bearing monomer, with 3-mercaptopropylglycine successfully replacing cysteamine. Internal thiols did not interfere with ligation selectivity. Isolated yields following purification reached 32 percent for model sequences. Radical desulfurization successfully converted the ligation-site thiol to an N-ethylglycine residue, demonstrating that the method can produce native peptoid sequences without residual functional groups. The chemistry also enabled intramolecular cyclization of a linear hexamer bearing both termini, achieving near-quantitative conversion to the macrocycle.
The team applied NCL to synthesize functional peptoid nanomaterials, ligating charged anchor blocks to hydrophilic loop cassettes to create 38-mer sequences capable of self-assembling into two-dimensional nanosheets at the air-water interface. Sequential ligation of three fragments demonstrated modular construction, where stable anchor blocks can be stored and combined with diverse loop sequences to generate libraries of functional nanosheets. Fluorescence microscopy confirmed that ligated products assembled into micron-scale sheets indistinguishable from those synthesized by conventional routes. One unexpected challenge emerged: peptoids bearing N-terminal amine side chains formed N-nitrosamines during hydrazide oxidation, adding 29 daltons to the product mass. Redesigning the charge orientation of nanosheet sequences eliminated this side reaction.
This work provides peptoid chemists with the first convergent ligation method that preserves native backbone connectivity. Unlike previous conjugation strategies relying on triazoles, disulfides, or other non-native linkages, NCL produces only tertiary amide bonds at ligation sites. The modular approach enables incorporation of low-yielding monomers into long sequences by confining them to short, purifiable fragments. Sequential ligation strategies promise accelerated discovery of functional peptoid macromolecules, including structures with defined tertiary architecture and biomimetic properties previously inaccessible due to synthetic limitations. The method opens routes to proteomimetic systems that rival natural biopolymers in complexity and function.