Hydrogels hold enormous promise for biomedical applications ranging from drug delivery and wound dressings to tissue modeling and soft robotics, yet their high water content leaves them inherently soft and fragile. These limited mechanical properties constrain their use in demanding contexts such as cartilage replacement, vascular grafts, and injectable therapeutics. Biological tissues solve this problem through fibrous proteins like collagen and actin, which form highly extended networks that stiffen and toughen the surrounding matrix. Replicating this strategy with synthetic materials has remained a persistent challenge, since most synthetic polymers curl into random coils rather than assembling into the long, rigid fibers that nature uses to bear mechanical loads.
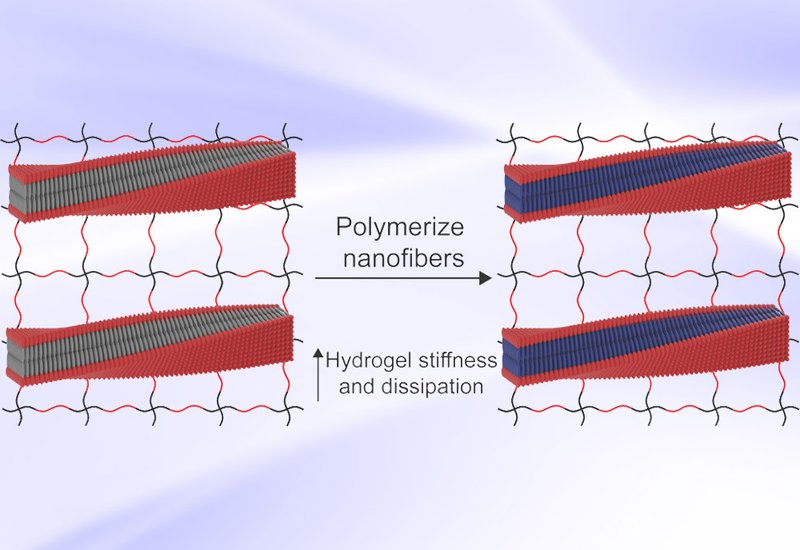
Researchers in the Pashuck Group at Lehigh University, published in the Journal of the American Chemical Society, developed a modular approach that harnesses self-assembling peptides to direct covalent polymerization inside hydrogels. The team designed a series of diacetylene peptide amphiphiles, molecules that combine a short peptide sequence with a polymerizable diacetylene tail derived from 10,12-pentacosadiynoic acid. These DA-PAs dissolve readily in water and spontaneously organize into high aspect ratio nanofibers through hydrogen bonding and hydrophobic interactions, forming structures comparable in scale to natural fibrous proteins. Upon UV irradiation, the diacetylene groups undergo topotactic polymerization, a reaction that occurs within the highly ordered lattice to form fully extended covalent polymer chains along the nanofiber axis without disrupting the assembled structure.
By systematically varying the peptide sequence, the team discovered that even single amino acid changes dramatically altered both nanofiber morphology and polymerization efficiency. Circular dichroism and transmission electron microscopy revealed that sequences forming thin, high aspect ratio nanofibers with well-ordered molecular packing polymerized most effectively, while those producing flat sheets or short fibrils showed negligible polymerization. A sequence designated V2A2E3, containing two valines, two alanines, and three glutamic acids, emerged as the clear standout. This peptide formed thin, high aspect ratio nanofibers and polymerized far more efficiently than all other candidates. Incorporating V2A2E3 into poly(ethylene glycol) hydrogels and triggering polymerization boosted stiffness 200-fold and viscous energy dissipation over 1,000-fold compared to PEG gels alone. The team further showed that the chemistry of the PEG cross-linker plays a critical role: positively charged peptide cross-linkers, which form electrostatic bonds with the negatively charged DA-PA nanofibers, produced gels nearly ten times stiffer than those made with neutral PEG cross-linkers. These interfacial ionic interactions act as sacrificial bonds that dissipate energy during deformation, mimicking strategies found in biological tissues. Cyclic strain experiments confirmed that these hydrogels recover more than 90% of their original stiffness after repeated high-strain episodes when peptide cross-linkers maintain ionic contact with the nanofiber network. To demonstrate broad applicability, the researchers added V2A2E3 DA-PAs to alginate hydrogels and achieved an almost 20-fold increase in stiffness, while gelatin hydrogels saw storage modulus gains exceeding 200-fold.
This work establishes a versatile, sequence-driven toolkit for engineering hydrogel mechanics from the bottom up. Because DA-PAs dissolve in water before polymerization, they can integrate into virtually any existing hydrogel system without complex fabrication steps, opening pathways to mechanically robust materials for cartilage repair, vascular grafts, flexible bioelectronic sensors, and injectable therapeutics. The study also offers fundamental insight into how hierarchical self-assembly, precise molecular control, and tunable interfacial interactions can bridge the performance gap between synthetic hydrogels and the remarkable mechanical sophistication of living tissues.