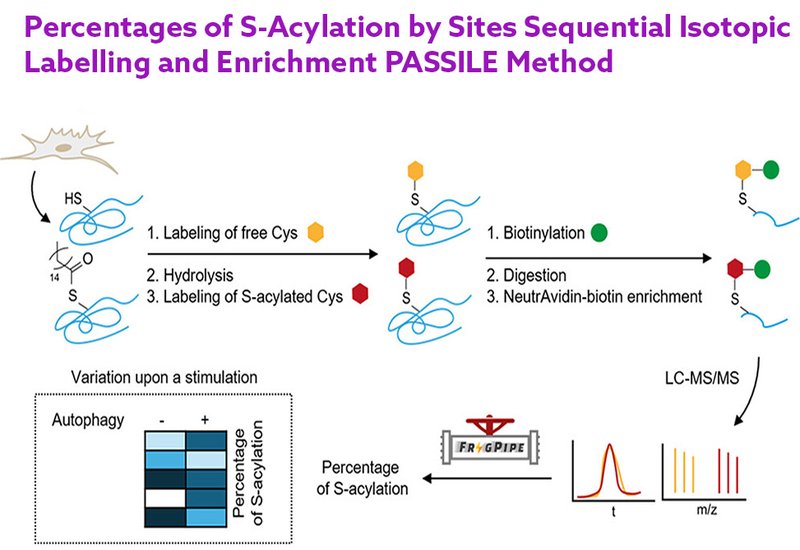
Researchers in the Emmanuelle Thinon Group at the University of Bordeaux, published in ACS Chemical Biology as part of the special issue "Lipids and Lipidation," have developed a chemical proteomics method that quantifies the percentage of S-acylation at individual cysteine residues across the proteome. The workflow, termed PASSILE for percentages of S-acylation by sites sequential isotopic labeling and enrichment, addresses a critical gap in understanding this dynamic lipid modification by enabling site-specific quantification independent of protein expression changes.
S-acylation involves the reversible attachment of fatty acids to cysteine residues through thioester bonds, with palmitic acid being the most common modification. This hydrophobic modification dramatically affects protein membrane association, localization, trafficking, and stability. The ZDHHC family of acyltransferases catalyzes the addition while serine hydrolases including APT1/2 and ABHD proteins remove the modification, creating a dynamic regulatory system. Aberrant S-acylation has been linked to cancers, viral infections, and neurodegenerative disorders. Existing mass spectrometry methods can identify S-acylated proteins but struggle to quantify the fraction of any given cysteine that carries the modification at a particular time. Relative comparisons between samples cannot distinguish whether apparent changes reflect altered S-acylation levels or simply changes in total protein abundance. The team adapted isotopic labeling strategies previously used to quantify cysteine oxidation, hypothesizing that sequential labeling of free and S-acylated cysteines with isotopically distinct probes could enable precise quantification of S-acylation stoichiometry.
The PASSILE workflow labels free cysteines with a light isotope-containing alkyne probe, then cleaves thioester-linked fatty acids with hydroxylamine and labels the newly exposed thiols with a heavy isotope version of the same probe. Copper-catalyzed click chemistry conjugates all labeled cysteines to a biotin capture reagent, enabling enrichment on NeutrAvidin beads after protein digestion. The ratio of heavy to light labeling at each cysteine directly reports its S-acylation percentage. The team systematically compared two alkyne-iodoacetamide probe sets with four different azido-biotin capture reagents, finding that the combination of IAA-Bn probes with a dialkoxydiphenylsilane-cleavable linker reagent yielded the highest number of quantified sites. Extensive optimization of sample preparation, including cleanup methods between labeling steps and digestion conditions, proved essential. Integration of high-field asymmetric waveform ion mobility spectrometry with liquid chromatography-tandem mass spectrometry further improved detection of labeled peptides. A custom FragPipe workflow and analysis script enabled calculation of S-acylation percentages from the quantified heavy and light ion intensities.
Applied to HeLa cell lysates, the optimized PASSILE method quantified over 17,000 unique cysteines in biological triplicates, representing improved coverage of the human cysteinome compared to previous oxidation-focused methods. Among sites showing S-acylation percentages above 10%, two-thirds belonged to proteins previously identified in palmitoyl-proteome datasets catalogued in SwissPalm. The team then applied PASSILE to identify dynamic S-acylation changes during autophagy, the cellular recycling process. Nutrient starvation induced significant changes at numerous sites, including increased S-acylation of Cys386 in ALDH2 and Cys26 in RAB14, both proteins with established roles in autophagy pathways. These proteins had been identified as S-acylated in previous studies, but the specific modified cysteines had not been validated. The method opens new avenues for investigating how dynamic S-acylation modulates protein function, though the authors note that coverage of membrane protein sites near transmembrane domains remains challenging due to the hydrophobicity of the resulting tryptic peptides. Future refinements including automation, alternative proteases, and improved capture reagents could further expand the accessible cysteinome.