Unnatural amino acids are indispensable tools in protein engineering, drug discovery, and diagnostics. Yet synthesizing them efficiently and in enantiopure form remains a persistent bottleneck. Traditional routes involve multiple steps: building the amino acid skeleton, installing a protecting group on the nitrogen, activating the carboxylic acid for coupling, and verifying that chirality survives each transformation. Each additional step costs time, reduces yield, and introduces opportunities for racemization. An ideal method would deliver unnatural amino acids that are shelf-stable, enantiopure, and ready to plug directly into a peptide chain without further modification.
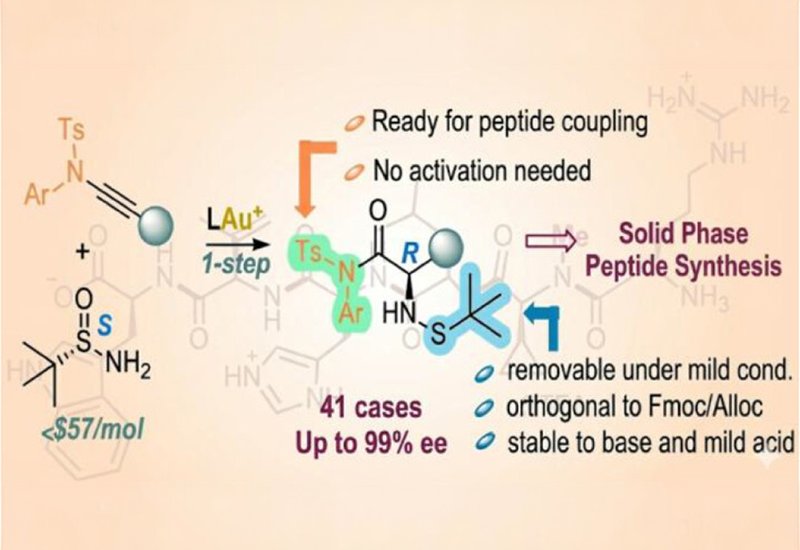
Researchers in the Zhang Group at the University of California, Santa Barbara, published in the Journal of the American Chemical Society, developed a concise two-step synthesis that converts readily available terminal alkynes into diverse unnatural amino acid building blocks primed for immediate use in peptide assembly. The strategy pairs mild gold(I) catalysis with inexpensive, commercially available chiral tert-butylsulfinamide. In the first step, a terminal alkyne is coupled to a sulfonamide to form an ynamide substrate. In the second step, a cationic gold(I) catalyst activates the ynamide toward addition of the chiral sulfinamide, generating a key intermediate with exclusive Z-geometry. This geometric control, achieved through gold-catalyzed anti-addition, creates a more congested transition state that delivers exceptional stereoselectivity. Using BrettPhos as the supporting ligand and NaBARF as the counterion at 50 degrees Celsius, the reaction routinely achieves 95% enantiomeric excess or higher, and simple recrystallization upgrades the products to enantiopurity.
What makes these building blocks particularly valuable for peptide chemists is their dual functionality. Each product carries a novel tert-butylsulfenyl group as the nitrogen protecting group and a mildly activated carboxylic acid moiety, meaning it can enter a peptide coupling reaction without additional activation reagents. The protecting group is stable under basic conditions and standard palladium-catalyzed deprotection protocols but can be cleanly removed in minutes using either trifluoroacetic acid or, for sequences requiring acid-sensitive protecting groups, pentafluorothiophenol. The team demonstrated that potassium oxymate facilitates efficient amide bond formation at ambient temperature, typically completing within three hours. Crucially, this coupling protocol produced negligible to no epimerization, even with notoriously racemization-prone residues like phenylglycine. Microwave-assisted coupling at 80 degrees Celsius achieved full conversion in just six minutes with less than 0.1% epimerization. The substrate scope spans more than 40 examples, including alkyl, aryl, heteroaryl, and alkenyl side chains, as well as functionalized derivatives bearing chloro, azido, cyano, and protected hydroxyl groups. N-methyl variants, which are valuable for modulating peptide bioactivity and oral bioavailability, were also accessible through use of N-methyl-tert-butylsulfinamide. The team validated both solution-phase and solid-phase peptide synthesis, assembling peptides up to six residues long and demonstrating seamless compatibility with standard Fmoc-based Oxyma/DIC coupling protocols.
This methodology addresses a longstanding gap between amino acid synthesis and peptide assembly by eliminating the protecting group and activation steps that traditionally bridge the two. The bench stability, broad scope, and epimerization-free coupling of these building blocks make them attractive for laboratories working on peptide therapeutics, peptidomimetics, and structure-activity relationship studies where rapid access to diverse unnatural residues is essential.