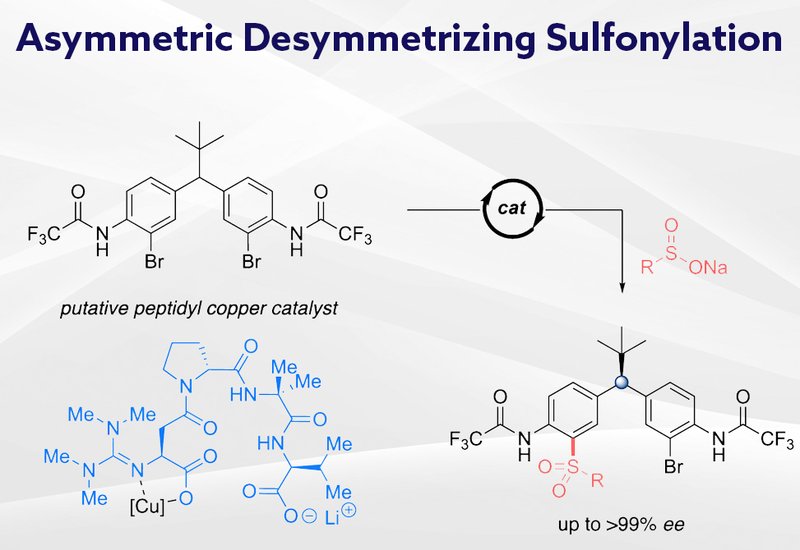
Researchers in the Miller Group at Yale University, published in the Journal of the American Chemical Society, have developed guanidinylated peptide ligands that enable copper-catalyzed asymmetric carbon-sulfur bond formation with remote stereocontrol. The chemistry achieves desymmetrization of diarylmethanes through cross-coupling with organosulfinates, installing sulfone groups while establishing a stereogenic center four atoms away from the newly formed bond. This work extends the utility of peptide-based asymmetric catalysis to a bond-forming process that had not previously been accomplished with remote stereoinduction.
Diarylmethane scaffolds appear throughout medicinal chemistry, making asymmetric access to these structures highly valuable. While peptide-based organocatalysts have proven effective for acylation and bromination reactions on diarylmethanes, extending peptide-mediated asymmetric catalysis to cross-coupling reactions presents distinct challenges. The Miller lab previously demonstrated that guanidinylated peptides serve as effective ligands for copper-catalyzed desymmetrizing cross-couplings forming carbon-carbon, carbon-oxygen, carbon-nitrogen, and carbon-iodine bonds. Sulfone-containing compounds constitute important scaffolds in pharmaceuticals, agrochemicals, and materials, yet catalytic enantioselective construction of carbon-sulfur linkages with remote stereocontrol had not been reported. The team hypothesized that sulfonylative desymmetrization might be achievable through peptidyl copper complexes engaging organosulfinate nucleophiles.
Initial experiments with L-proline as a ligand produced the mono-sulfonylated product in 20% yield with modest enantioselectivity, confirming that the transformation was feasible. Systematic optimization revealed acetonitrile as the optimal solvent and identified that lowering the reaction temperature from 90°C to 50°C dramatically improved both yield and selectivity. The ortho-trifluoroacetamide substituent on the diarylmethane substrate proved essential, accelerating the coupling under mild conditions while suppressing hydrolysis pathways. Screening guanidinylated peptide ligands showed that a tetrameric sequence delivered the best results, affording the tosylated product in 72% yield with 92% enantiomeric excess. The optimized conditions tolerated aryl, alkyl, and heteroaryl sulfinates, with pyridyl sulfones obtained in yields exceeding 57% and enantioselectivities reaching 96%. Increasing steric bulk on alkyl sulfinates slightly diminished both yield and selectivity, suggesting that reductive elimination and the geometry of catalytic intermediates are sensitive to nucleophile size. Mechanistic studies revealed that secondary kinetic resolution operates during the catalysis, further enriching the optical purity of products. Extending the reaction time from 2 to 4 hours elevated the enantiomeric excess of one substrate from 95% to greater than 99%, providing an operationally simple method to enhance selectivity for intrinsically less selective cases.
The catalytic system requires both base and the trifluoroacetamide directing group, and countercations including cesium, potassium, and sodium influence the asymmetric induction. The team proposes that an intricate multivalent substrate-copper-peptide complex forms, organized through ensembles of noncovalent interactions including ionic contacts and cation-π interactions. The peptide ligand likely adopts a β-turn-biased architecture stabilized by intramolecular hydrogen bonding, while the bulky substituent at the prochiral carbon center of the diarylmethane organizes the substrate geometry for effective molecular recognition. The synthetic utility of this chemistry was demonstrated through one-pot sequential functionalization, where sulfonylation followed by etherification with 4-methoxyphenol delivered an unsymmetrically bis-functionalized diarylmethane in 94% enantiomeric excess. Site-selective sulfonylation of polybrominated substrates occurred exclusively at bromides ortho to the directing group, and the reaction scaled to 1 mmol without loss of stereoselectivity. This work establishes peptide-based ligands as versatile tools for asymmetric carbon-sulfur bond construction, expanding the scope of transformations amenable to biomimetic catalyst design while providing practical access to enantioenriched sulfone-containing diarylmethane building blocks.