Bicyclic peptides occupy a sweet spot in drug discovery. Their constrained architectures resist proteolytic degradation and bind targets with antibody-like specificity, yet they remain small enough for potential oral delivery. Building libraries of these molecules for screening presents a challenge, however. The most powerful selection platform, mRNA display, can search through trillions of sequences but demands chemistry gentle enough to preserve the fragile genetic readout. A team led by Hiroaki Suga at the University of Tokyo has now developed a ribosomal route to thioisoindole-bridged bicyclic peptides that threads this needle, enabling genetically encoded access to a topologically defined scaffold class previously inaccessible to display technologies.
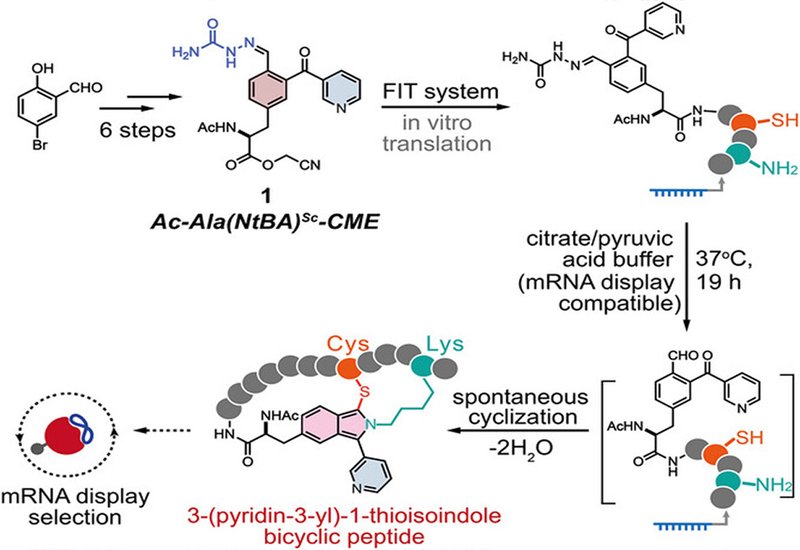
The chemistry draws inspiration from ortho-phthalaldehyde, which reacts with lysine to form an iminium intermediate that cysteine then traps, yielding a thioisoindole bridge. While elegant, raw phthalaldehyde proves too reactive for translation systems: it would attack free amino acids and proteins in the cellular extract before the desired peptide even forms. The researchers solved this reactivity problem through careful molecular design. They synthesized a phenylalanine derivative bearing a 2-nicotinoylbenzaldehyde group with the aldehyde masked as a semicarbazone. The nicotinoyl substituent tunes down electrophilicity compared to simple phthalaldehyde, while the semicarbazone protection survives translation conditions but cleaves under mild acid.
The team optimized a convergent synthesis featuring a Negishi coupling between an iodophenylalanine fragment and a brominated nicotinoylbenzaldehyde, achieving 67% yield without protecting the reactive carbonyl groups. When tested as a flexizyme substrate for tRNA charging, the optimized compound outperformed even the commonly used chloroacetyl-tryptophan initiator, reaching near-quantitative aminoacylation at 25 mM. The charged tRNA then entered the flexible in vitro translation system, where it initiated synthesis from the AUG start codon, placing the masked cyclization handle at the peptide N-terminus.
After translation, adding citrate buffer at pH 4 containing pyruvic acid triggered semicarbazone removal. The liberated aldehyde spontaneously attacked a lysine side chain within the same peptide, and the resulting iminium underwent intramolecular capture by a cysteine thiol, forging the thioisoindole bridge and closing the bicyclic topology. The researchers tested 14 different peptide sequences varying in length, ring size, and the relative positions of the cysteine and lysine residues. Twelve constructs cyclized with 60 to 98% conversion; only sequences placing acidic residues immediately adjacent to the cyclization sites showed reduced efficiency. Ring sizes ranged from small to large without compromising the reaction, demonstrating broad sequence tolerance.
Crucially, the deprotection conditions preserved mRNA integrity. Citrate buffer, commonly used for RNA storage, protected the genetic template from base hydrolysis during the overnight incubation. Both qPCR recovery and gel electrophoresis confirmed that neither mRNA nor mRNA/cDNA hybrids suffered detectable degradation. This compatibility means the entire workflow can integrate directly with the RaPID selection system, where mRNA-displayed peptide libraries undergo iterative rounds of target binding and amplification to enrich high-affinity sequences.
This work opens a new corridor in the chemical space accessible to genetically encoded peptide libraries. Thioisoindole bridges create rigid, metabolically stable architectures distinct from the disulfide and thioether linkages that dominate current bicyclic peptide selections. The Suga lab's strategy requires no exogenous crosslinking reagents and proceeds intramolecularly, avoiding the off-target modifications that plague intermolecular approaches. For drug hunters seeking constrained peptide scaffolds against challenging targets, ribosomal access to this underexplored topology represents a significant expansion of the discovery toolkit.