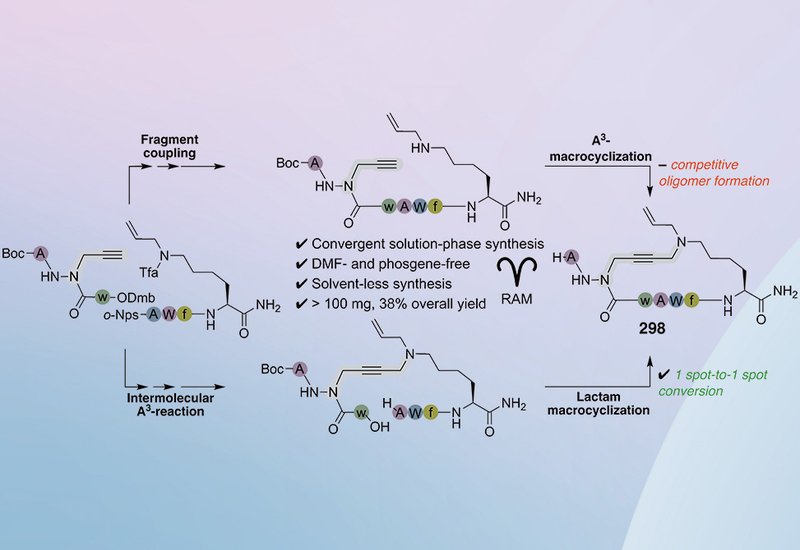
The cluster of differentiation-36 receptor, CD36, sits at the crossroads of metabolic, vascular, and immunological signaling, and its dysregulation has been linked to atherosclerosis, diabetes, non-alcoholic fatty liver disease, and cancer. Macrocyclic azapeptide 298, discovered through copper-catalyzed aldehyde–alkyne–amine A3 chemistry in solid-phase peptide synthesis, SPPS, is a potent CD36 modulator with demonstrated anti-inflammatory and cardioprotective activity in multiple mouse disease models. The SPPS route delivered only milligram quantities after preparative HPLC purification and relied on reprotoxic DMF and phosgene, making it poorly suited for the larger material demands of broader preclinical work. A scalable, greener synthesis was needed.
A team of researchers led by doctoral candidate Nassim Maarouf-Mesli in the Lubell Group at Université de Montréal, published in J. Org. Chem., designed two convergent solution-phase routes that each assemble 298 from shared, orthogonally protected aza-tripeptide and tetrapeptide building blocks. Both fragments were constructed using resonant acoustic mixing, RAM, for amide bond formation and semicarbazide assembly at high concentrations in dimethoxyethane, a greener solvent, avoiding both DMF and phosgene. The two routes diverged at the macrocyclization step: one employed an intramolecular A3-reaction to close the ring between the azaPra and Lys side chains, mirroring the SPPS strategy, while the other used intermolecular A3-chemistry to forge the same side-chain linkage in the linear aza-isopeptide precursor, followed by lactam formation between D-Trp and Ala to close the ring.
At small scale, the intramolecular A3-macrocyclization of the linear azapeptide gave greater than 90% conversion in dilute DMSO/water. Scaling to 20–100 mg revealed a sensitivity to oligomer formation, and varying the copper salt or formaldehyde stoichiometry did not resolve the problem. The optimized A3-macrocyclization route ultimately delivered only 2 mg of 298 at 5% yield over two steps, even after HPLC purification. By contrast, lactam formation on the aza-isopeptide proceeded cleanly. Standard carbodiimide coupling with EDCI gave low conversion even after 48 hours, but HATU and HOAt in 1:9 MeCN/1,2-dichloroethane at 1.50 mM achieved spot-to-spot conversion on TLC and HPLC. On a 250 mg scale, this step delivered the protected macrocycle in 86% yield at 96% purity after aqueous workup and trituration.
Final Boc deprotection of 130 mg of the protected macrocycle with a TFA/triisopropylsilane/water/thioanisole cocktail, followed by serial precipitations, furnished 100 mg of cyclic azapeptide 298 in 84% yield and 99% purity without any chromatography. HPLC co-injection confirmed the solution-phase material, 298 LPPS, was identical to the earlier SPPS material. Biological evaluation showed that 298 LPPS and 298 SPPS carried comparable CD36 binding affinities of 0.279 and 0.201 µM, respectively, and produced similar inhibition profiles on TNF-α and CCL2 secretion from RAW264.7 macrophages stimulated with a TLR2 agonist.
This work establishes a practical, RAM-assisted convergent solution-phase route to a complex macrocyclic azapeptide without preparative chromatography, phosgene, or DMF. The key design insight is that placing the A3-reaction in the intermolecular fragment-joining step, rather than the macrocyclization step, circumvents the oligomerization problem that plagues high-dilution ring-closing A3-chemistry. The 100 mg scale achieved here is sufficient to support expanded pharmacological and pharmacokinetic profiling of 298, and the protecting-group strategy and RAM-assisted coupling conditions are applicable to other cyclic azapeptide targets.